基因工程是以分子生物学和微生物学的现代方法为手段,将不同来源的基因按预先设计的蓝图,在体外构建杂种DNA分子,然后导入活细胞,以改变生物原有的遗传特性、获得新品种、生产新产品。本文提供几篇有关基因工程的论文优秀范文,供大家学习。
有关基因工程的论文一:
[摘要]目的构建含有人纤维蛋白原基因的毕赤酵母表达系统,实现胞外高效分泌表达。方法全基因合成人纤维蛋白原3个基因FGA、FGB、FGG,构建表达载体pGAPZαA-FGB-FGG-FGA-AOX1,线性化后电转化导入毕赤酵母菌株SMD1168H,抗性筛选获得阳性克隆。发酵液经SDS-PAGE确定蛋白表达部位,ELISA检测目的蛋白表达量。表达产物超滤浓缩后利用AKTA蛋白纯化系统进行分离纯化,Westernblot检测蛋白表达情况并对纯化产物进行生物学活性测定。结果基因工程菌株摇瓶培养上清液表达量约15mg/L,生物学活性分析重组蛋白具有凝集活性。结论成功获得了高效分泌表达重组人纤维蛋白原的毕赤酵母菌株,且分离纯化的蛋白具有生物凝集活性。
[关键词]重组人纤维蛋白原;毕赤酵母;分泌表达;分离纯化
目前世界卫生组织确认的凝血因子共13个,大多由肝脏产生,正常情况下,所有凝血因子都处于无活性状态,以无活性酶原形式存在,当某一凝血因子被激活后,可使许多凝血因子按一定的次序先后被激活,逐级放大,直到纤维蛋白形成,血液发生凝固。纤维蛋白原(fibrinogen,Fg),即凝血因子Ι,是参与血液凝固的重要凝血因子,血浆中含量高达2000~4000mg/L[1],其分子量340kDa,由完全相同的2个亚基组成共价二聚体,每个亚基含有α(63.5kDa)、β(56kDa)、γ(47kDa)3条肽链[2],分别由4号染号体(4q28-30)上的3个独立的基因FGA、FGB、FGG编码形成,在肝脏中由独立的核糖体合成其前体蛋白,再经过内质网和高尔基体完成蛋白的组装,各肽链彼此通过二硫键相互连接形成Fg单体。2个单体通过3对链间二硫键连接形成对称性二聚体,即Fg。
纤维蛋白原参与凝血过程的机理:当血液中的凝血酶原被激活时,凝血酶作用于纤维蛋白原,切断纤维蛋白原α链N末端区域和β链N末端区域,α链的解离有利于纤维蛋白网络呈线性增长,β链的解离有利于纤维蛋白网络呈侧向增长,最终纤维蛋白单体以错综重叠状态侧向聚合缔结为网络构成的血凝块[3]。凝血初期非共价键结合可溶性的血凝块,然后在体内凝血因子ⅩⅢ(谷氨酰胺转移酶,又称纤维蛋白稳定因子)和Ca2+作用下,纤维蛋白单体通过ε-氨基-γ-谷氨酰基连接,形成不溶性的稳定血凝块[4],从而发挥凝血和生理性止血功能。另外,纤维蛋白原还参与体内血小板聚集、纤溶调节、动脉硬化的发生发展、组织损伤及修复等一系列病理和生理过程[5-8]。
医药级纤维蛋白原提取于人体血液,生产成本高昂且难以避免血源性传染病(如乙肝和艾滋病等)的传播风险,影响其市场应用及推广。随着分子生物学发展,采用基因工程方法高效表达安全、可靠的人纤维蛋白原成为研究热点。2011年,黄星等[9]将人纤维蛋白原基因在大肠杆菌BL21菌株中进行诱导表达,发现人纤维蛋白原3个亚基不能共表达,即纤维蛋白原的β、γ链能用pET系列载体获得表达,而α链却未能获得表达。大肠杆菌等原核生物表达系统还存在目的蛋白无法正确折叠修饰等空间结构问题,影响纤维蛋白原生物活性。动物细胞表达系统中,虽然可以通过哺乳动物乳腺生物反应器获得特异性表达具活性的纤维蛋白原,但存在投入成本高、基因打靶的位置效应导致基因整合具随机性、重组蛋白与天然蛋白修饰加工仍存在差异等问题,难以实现规模化生产[10-11]。酵母表达系统可以克服大肠杆菌和动物细胞表达系统的不足,通过高密度发酵实现工业化生产,蛋白质翻译后的修饰加工功能更加完善,蛋白产物更接近天然蛋白质功能。本研究拟通过构建重组人纤维蛋白原酵母表达载体获得高效分泌表达的毕赤酵母菌株,分离纯化具生物活性蛋白,为进一步开发有效的药用纤维蛋白原奠定基础。
1、材料与方法
1.1、材料
1.1.1、质粒与菌株:质粒pGAPZαA(Invitrogen),含有GAP启动子和Zeocin抗性基因,作为表达载体;质粒pPIC9K,仅用于AOX1基因的PCR扩增,并将该基因连接入pGAPZαA表达载体,利用AOX1作为线性化位点进行同源重组,以上质粒均为本实验室保藏。菌株以毕赤酵母(Pichiapastoris)SMD1168H作为表达宿主,为本实验室保藏。
1.1.2培养基:低盐LB培养基(g/L):蛋白胨10,酵母粉5,氯化钠5,pH7.5;YPD培养基(g/L):胰蛋白胨20,酵母粉10,葡萄糖20,固体培养基含1.5%琼脂粉。
1.1.3、试剂:限制性内切酶、T4DNA连接酶等购自NEB公司、Taq酶、DNAMarker、蛋白分子量Marker等购自Fermentas公司,抗生素Zeocin购自Invitrogen公司,质粒抽提试剂盒、PCR产物纯化试剂盒、DNA胶回收试剂盒购自Omega公司,超滤管购自Millipore公司,层析柱填料SephacrylTMS-100HR购自GE公司,ELISA试剂盒购自Bio-Swamp公司,鼠抗人单克隆抗体(α、β、γ)、辣根过氧化物酶偶联的羊抗鼠IgG均购自SantaCruz公司,DAB显色试剂盒购自Tiangen公司。其他试剂均为市售分析纯。
1.1.4、仪器:摇床(伊孚森生物技术中国有限公司)、PCR仪(Eppendorf)、离心机(Thermo)、电转化仪(Biorad)、酶标仪(Tecan)、蛋白电泳设备(Biorad)、AKTA蛋白分离纯化仪(AKTAExplorer)、生物样品均质器(Bertin)。
1.2、方法
1.2.1、pGAPZαA-FGB-FGG-FGA-AOX1重组质粒的构建
NCBI数据库检索人纤维蛋白原FGA、FGB、FGG基因序列(GeneID分别为2243、2244、2266),根据宿主菌株毕赤酵母密码子偏好性进行序列优化,并设计合适的酶切位点,对含GAP启动子和AOX1TT终止子表达框的全基因序列合成,重组质粒载体如图1所示,具体步骤如下:
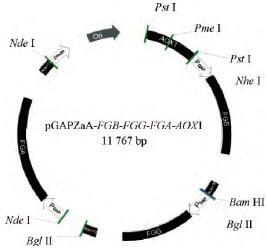
图1 重组人纤维蛋白原酵母表达载体的构建图
①限制性内切酶NheⅠ、BamHⅠ分别双酶切FGB基因和pGAPZαA质粒,经纯化连接转化E.coliTop10感受态细胞,于25μg/mLZeocin低盐LB固体培养基筛选阳性转化子,获得pGAPZαA-FGB重组质粒。
②限制性内切酶BglⅡ分别单酶切FGG基因和pGAPZαA-FGB质粒,载体脱磷酸化并纯化,连接转化E.coliTop10感受态细胞,抗性筛选获得pGAPZαA-FGB-FGG重组质粒。
③限制性内切酶NdeⅠ分别单酶切FGA基因和pGAPZαA-FGB-FGG质粒,载体脱磷酸化并纯化,连接转化E.coliTop10感受态细胞,抗性筛选获得pGAPZαA-FGB-FGG-FGA重组质粒。
④以pPIC9K质粒为模板PCR扩增AOX1基因,PstⅠ单酶切AOX1基因和pGAPZαA-FGB-FGG-FGA重组质粒,载体脱磷酸化并纯化,连接转化E.coliTop10感受态细胞,抗性筛选获得pGAPZαA-FGB-FGG-FGA-AOX1重组质粒。
每一步重组质粒构建均经生物公司测序正确后,再进行下一个目的片段的连接。
1.2.2、毕赤酵母的电转化及PCR扩增验证:PmeⅠ单酶切线性化重组质粒pGAPZαA-FGB-FGG-FGA-AOX1,胶回收纯化,按《毕赤酵母表达操作手册》[12]方法电转化导入毕赤酵母SMD1168H感受态细胞,电转化条件:1.5kV,200Ω,25μF。电转化后酵母细胞涂布于含1μg/mLZeocin抗性平板,28℃培养2d,筛选阳性转化子,以破壁重组子为模板,PCR扩增检测重组载体的FGA、FGB、FGG基因验证。
1.2.3、重组人纤维蛋白原的表达:挑取单克隆于YPD培养基(含1μg/mLZeocin),28℃,220r/min培养60h。采用生物样品均质器对发酵液及菌体破碎,均质速度6500r/min,3个循环,每个循环运行时间为30s,循环间隔30s。参考《精编分子生物学实验指南》[13]对全细胞破碎液、菌体沉淀破碎液及发酵上清液进行SDS-PAGE电泳检测,确定目的蛋白的表达部位,ELISA试剂盒检测确定目的蛋白的表达量。
1.2.4、重组人纤维蛋白原的Westernblot检测:发酵上清液经30kDa超滤管浓缩,AKTA纯化系统分离,收集蛋白吸收峰值处洗脱液进行二次超滤浓缩获得纯化蛋白。纯化条件:分子筛层析柱ColumnXK16,凝胶填料为SephacrylTMS-100HR,流速0.5mL/min,上样量5mL。纯化蛋白分别以小鼠单克隆一抗(α、β、γ)(稀释比例1∶1000),羊抗鼠IgG-HRP作为二抗(稀释比例1∶10000)进行Westernblot,抗体孵育结束后洗膜3次,DAB显色试剂盒显色,具体操作方法参考《精编分子生物学实验指南》[13]。
1.2.5、重组人纤维蛋白原的生物活性测定:AKTA纯化蛋白洗脱液4℃透析过夜(20mMHEPES,pH7.4,150mMNaCl,5mMεACA,1mMCaCl2),添加外源0.1U/mL凝血酶、0.02U/mL凝血因子ⅩⅢ,37℃条件下参照2015年版中国药典方法[14]检测纤维蛋白原凝集活性。





