�����ο��������ݿ��Ƿ�ȫ��������ļ����Ӱ��ϴ������������ַ��������ݿ�������ۺϱȽϣ�������3.
����
����
����
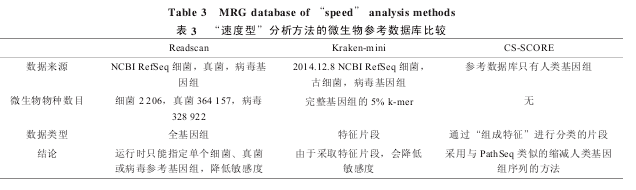
����“�ٶ���”��ⷽ���������������е��ٶ�����������Ż������� B������ Kraken �ľ������ݿⷨ��CS-SCORE �Ļ��� cs-score �ıȶԷ����ȣ����ںܴ�̶��ϼ����˱ȶԺ������̡������ٶȵ����ӻ�ɱ���ش������ȵ��½������罵�Ͳο��������С�����Ӽ���ٶȣ����ǻή�ͱȶԹ�������ȷƥ���reads �������Ӷ��������жȣ����Ӳο��������С�ܹ���߽����ȷ�ԣ������������������ӣ������ٶ�Ҳ����Ӧ���͡�Ŀǰ���µļ�ⷽ��һ���ṩ������ģʽ����һ��ģʽΪֻ������������ MRG ���бȶԵĿ���ģʽ�����ŵ����ڼ���ٶȿ죬ȱ�����������MRG ��ȱʧ�����У���һ��Ϊ���������������еķ�ʽ���ŵ����ܹ����δ֪�������У�ȱ���Ǽ���ٶȽϵ͡��û����Ը�������ѡ���ʺ��Լ��ļ��ģʽ����Ҳ������������ٶȺ;���֮�����ƽ�����ѷ�����
����
����
4�ܽ���չ����
����
�������� NGS ���������������ݷ���������������Ԥ�����������������ȸߡ��ܹ����δ֪��������ص㣬Ϊ�������غ�����ʳƷ��ȫ�ṩ���µĽ�����������Ķ�Ŀǰ������ 12 �ֻ��� NGS�������ⷽ�������˼�Ҫ���ܺͱȽ��о����Ը�����ⷽ�����������̺����ݴ��������ֱ�����˻����Ϻ��Ż������ϵķ����������ݷ������ڷ��棬������NGS �����������ݷ���������Ϊ“������”,“������”,“�ٶ���”,“������”���������͡����Ż������棬������������ָ�꣺�ٶȡ������Լ��ĸ�Ӧ�����أ�������Դ����ϵ�ṹ���ܺĺͿ���չ�Է�����бȽϷ�����ͨ�����������ݷ�����������ʵ�ֵ��ܽ����ۣ�ϣ��Ϊ����ͼ������������о��ṩ�ο���ֵ��
����
������ʵ����Ҳ�Ը�������о�������һ����̽���������[48]��Դӷ����������м���δ֪��ԭ��������⣬��������������������̽����˷��������������������бȶ�����Bowtie2��BWA�� BLAST + �� MUMer[49];������ƴ������Velvet[50]�� SOAPdenovo[51]; �� �� �� �� �� ��BEDTools[52]�� MEGAN4[53]�� MAUVE[54]�� IGV[55]��Circos[56]�ȡ��ֱ�ӳ��没ԭ���⡢��ͻ�����µIJ�ԭ���⡢��ͬ��������Լ������µIJ�ԭ���⡢�����������ʱ�IJ�ԭ����ͻ����Ʒ�еIJ�ԭ���⼸�����棬��������������ϵͳ������ ���о������������������ָ���Ժ��ٴ���ԭ�����������������Ҷ��ǿ��[57]��Ե��ܹܽ�ʯ���������о����⣬ʹ�ú����������ֶΣ���15 λ�й����ܹܽ�ʯ���ߵĵ�֭��������ȫ���������ǹ������� 16 S �����������Ӳ�����������о�������13 ��֮ǰδ�����ĸ����鸲�Ƕȵĵ���ϸ������������뵨ʯ�γ�֭������صĻ����������⼼���ڼ������������ϵ�ʵ��Ӧ�á������[58]������“��Ӷ���”�ϵĻ��� IntelMIC�ĸ�ͨ�� DNA ���бȶԲ�������������DNA ���бȶ����� MICA[57]�����Ӷ��ų����������Ӳ���ܹ���ƣ��ܹ���ַ���MIC �IJ���DZ�������нӽ����Լ��ٱȵ���չ���ܡ�����Ĺ������ڴ�ϸ�����϶Ի���NGS ��������������������������ܹ��������ȣ���ⷽ������ϸ���ȣ��������������ϣ����ܹ�ʹ�˸������ط������ܽ�������Ĺ��̡�Ҷ��ǿ�Ĺ������ڴ�ͳ�����ⷽ���Ľ�һ����չ������ܹ���16 S �����ͻ���NGS �����������ϣ����ܹ�ʹ������ȷ������Ĺ����ܹ��ƶ����� NGS �������ⷽ�����ٶȸ��ŷ���չ����ϱ�ʵ���������о������ܹ������������о������ٽ������Ȿ�����ڿ����齨��������һ�������ۺ���������NGS ������ļ�����������������������ݺ�����������ͬͻ���ʵIJ������ݡ���ͬ��ģ�IJ��������Լ�����ʵ���ݽӽ���ģ�����ݣ����ѧ���ɹ���Ͷ��PDP 2017 ���ʻ��顣
����
����δ������NGS �������ⷽ���ķ�չ����������ٶȺ;��������ص��Ż�������֮�⣬���м�����������ڼ�����Դ���ܺġ���ϵ�ṹ�ȷ���Ҳ���������Ż��ռ䡣������Դ���棬ͨ������ο����ȷ����ܹ���Ч���������ڴ棻Ҳ����ͨ��ר��Ӳ�����ٿ���������������������ݷ��������Ĵ���ʱ�䣬���õ����������в������ϵ�ṹ���棬��ͨ���ϴ��������Ƽ��������������ʹ��ⷽ���и��ߵIJ���ϵͳ�����Եȡ���GPU��ARM���ܺĴ������Լ�FPGA���ܹ��Ӳ�ͬ����϶Լ�ⷽ�����м��ٻ�����������Ż���
����
�������˼��㼼��������Ż������\���Ĵ���Ҳ�ܸ������ⷽ���������¡�δ�����\���ķ�չʹ���������Ÿ�ͨ�����ͳɱ�������ȡ���ȵķ���չ��Ŀǰ�ѽӽ�ʵ�õĵ��������������г����������ص㣬һ��Ӧ�ý��Ἣ��ظ��������ⷽ�������̡������������ݷ�������������µ�Ҫ�������Ҫ�µ����ݷ��������ܹ���Ӧ�����������ٶȺ;����ϴﵽ���ߵı���Ϊ�����������ṩ�����١���ȷ�������ⷽ����
����
������ �� �� �ף�
����
����[1] Steingart K R, Henry M, Ng V, et al. Fluorescence versusconventional sputum smear microscopy for tuberculosis: asystematic review. The Lancet Infectious Diseases, 2006, 6 ��9����570-581.
����[2] Lemieux B, Aharoni A, Schena M. Overview of DNA chiptechnology. Molecular Breeding, 1998, 4��4���� 277-289.
����[3] Belgrader P, Benett W, Hadley D, et al. Rapid pathogen detectionusing a microchip PCR array instrument. Clinical Chemistry, 1998,44��10���� 2191-2194.
����[4] Call D R. Challenges and opportunities for pathogen detectionusing DNA microarrays. Critical Reviews in Microbiology, 2005,31��2���� 91-99.
����[5] Lazcka O, Del Campo F J, Munoz F X. Pathogen detection: aperspective of traditional methods and biosensors. Biosensors andBioelectronics, 2007, 22��7���� 1205-1217.
����[6] Schuster S C. Next-generation sequencing transforms today'sbiology. Nature, 2007, 200��8���� 16-18.
����[7] Barzon L, Lavezzo E, Costanzi G, et al. Next-generation sequencingtechnologies in diagnostic virology. Journal of Clinical Virology,2013, 58��2���� 346-350.
����[8] Reis-Filho J S. Next-generation sequencing. Breast CancerResearch, 2009, 11��3���� 1-8.
����[9] Metzker M L. Sequencing technologies-the next generation.Nature Reviews Genetics, 2010, 11��1���� 31-46.
����[10] Mandal P, Biswas A, Choi K, et al. Methods for rapid detection offoodborne pathogens: an overview. American Journal of FoodTechnology, 2011, 6��2���� 87-102.
����[11] Li R, Zhu H, Ruan J, et al. De novo assembly of human genomeswith massively parallel short read sequencing. Genome Research,2010, 20��2���� 265-272.
����[12] Li H, Homer N. A survey of sequence alignment algorithms fornext-generation sequencing. Briefings in Bioinformatics, 2010,11��5���� 473-483.
����[13] Mardis E R. Next-generation DNA sequencing methods. Annu RevGenomics Hum Genet, 2008, 9��3����87-402.
����[14] Wang D G, Fan J B, Siao C J, et al. Large-scale identification,mapping, and genotyping of single-nucleotide polymorphisms inthe human genome. Science, 1998, 280��5366���� 1077-1082.
����[15] Kostic A D, Ojesina A I, Pedamallu C S, et al. PathSeq: software toidentify or discover microbes by deep sequencing of human tissue.Nature Biotechnology, 2011, 29��5���� 393-396.
����[16] Bhaduri A, Qu K, Lee C S, et al. Rapid identification of non-humansequences in high-throughput sequencing datasets. Bioinformatics,2012, 28��8���� 1174-1175.
����[17] Borozan I, Wilson S, Blanchette P, et al. CaPSID: A bioinformaticsplatform for computational pathogen sequence identification inhuman genomes and transcriptomes. BMC Bioinformatics, 2012,13��1���� 157.
����[18] Chen Y, Yao H, Thompson E J, et al. VirusSeq: software to identifyviruses and their integration sites using next-generation sequencingof human cancer tissue. Bioinformatics, 2013, 29��2���� 266-267.
����[19] Wang Q, Jia P, Zhao Z. VirusFinder: software for efficient andaccurate detection of viruses and their integration sites in hostgenomes through next generation sequencing data. PloS One, 2013,8��5���� e64465.
����[20] Naeem R, Rashid M, Pain A. READSCAN: a fast and scalablepathogen discovery program with accurate genome relativeabundance estimation. Bioinformatics, 2013, 29��3���� 391-392.
����[21] Wood D E, Salzberg S L. Kraken: ultrafast metagenomic sequenceclassification using exact alignments. Genome Biol, 2014, 15 ��3����R46.
����[22] Naccache S N, Federman S, Veeraraghavan N, et al. Acloud-compatible bioinformatics pipeline for ultrarapid pathogenidentification from next-generation sequencing of clinical samples.Genome Research, 2014, 24��7���� 1180-1192.
����[23] Scheuch M, H�iper D, Beer M. RIEMS: a software pipeline forsensitive and comprehensive taxonomic classification of reads frommetagenomics datasets. BMC Bioinformatics, 2015, 16��1���� 1.
����[24] Kilianski A, Carcel P, Yao S, et al. Pathosphere. org: pathogendetection and characterization through a web-based, open sourceinformatics platform. BMC Bioinformatics, 2015, 16��1���� 1.
����[25] Haque M M, Bose T, Dutta A, et al. CS-SCORE: Rapididentification and removal of human genome contaminants frommetagenomic datasets. Genomics, 2015, 106��2���� 116-121.
����[26] Wang Q, Jia P, Zhao Z. VERSE: a novel approach to detect virusintegration in host genomes through reference genomecustomization. Genome Medicine, 2015, 7��1���� 1-9.
����[27] Li Y, Wang H, Nie K, et al. VIP: an integrated pipeline formetagenomics of virus identification and discovery. ScientificReports, 2016, 6: 23374· 67·��
����[28] Li H, Durbin R. Fast and accurate short read alignment withBurrows-Wheeler transform. Bioinformatics, 2009, 25 ��14���� 1754-1760.
����[29] Chen Y, Ye W, Zhang Y, et al. High speed BLASTN: anaccelerated MegaBLAST search tool. Nucleic Acids Research,2015, gkv784.
����[30] Altschul S F, Gish W, Miller W, et al. Basic local alignment searchtool. Journal of Molecular Biology, 1990, 215��3���� 403-410.
����[31] Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.Genome Biol, 2009, 10��3���� R25.
����[32] Kent W J. BLAT-the BLAST-like alignment tool. GenomeResearch, 2002, 12��4���� 656-664.
����[33] Lee W-P, Stromberg M P, Ward A, et al. MOSAIK: a hash-basedalgorithm for accurate next-generation sequencing short-readmapping. PloS One, 2014, 9��3���� e90581.
����[34] Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architectureand applications. BMC Bioinformatics, 2009, 10��1���� 1-9[35] Ponstingl H, Ning Z. SMALT-a new mapper for DNA sequencingreads. F1000 Posters, 2010, 1:313.
����[36] Zaharia M, Bolosky W J, Curtis K, et al. Faster and more accuratesequence alignment with SNAP. arXiv preprint arXiv:11115572,2011.
����[37] Ye Y, Choi J H, Tang H. RAPSearch: a fast protein similaritysearch tool for short reads. BMC Bioinformatics, 2011, 12��1���� 1.
����[38] Margulies M, Egholm M, Altman W E, et al. Genome sequencingin microfabricated high-density picolitre reactors. Nature, 2005,437��7057���� 376-380.
����[39] Langmead B, Salzberg S L. Fast gapped-read alignment withBowtie 2. Nature Methods, 2012, 9��4���� 357-359.
����[40] Li H, Durbin R. Fast and accurate short read alignment withBurrows-Wheeler transform. Bioinformatics, 2009, 25 ��14���� 1754-1760.
����[41] Bhatt A S, Manzo V E, Pedamallu C S, et al. Brief report: in searchof a candidate pathogen for giant cell arteritis: sequencing-basedcharacterization of the giant cell arteritis microbiome. Arthritis &Rheumatology, 2014, 66��7���� 1939-1944.
����[42] Hercus C. Novoalign. Selangor: Novocraft Technologies, 2012.
����[43] Zeitouni B, Boeva V, Janoueix-Lerosey I, et al. SVDetect: a tool toidentify genomic structural variations from paired-end andmate-pair sequencing data. Bioinformatics, 2010, 26 ��15���� 1895-1896.
����[44] Simpson J T, Wong K, Jackman S D, et al. ABySS: a parallelassembler for short read sequence data. Genome Research, 2009,19��6���� 1117-1123.
����[45] Treangen T J, Sommer D D, Angly F E, et al. Next generationsequence assembly with AMOS. Current Protocols inBioinformatics, 2011, 11��S33���� 11.18. 1-11.18. 18.
����[46] Morgulis A, Gertz E M, Sch��ffer A A, et al. A fast and symmetricDUST implementation to mask low-complexity DNA sequences.Journal of Computational Biology, 2006, 13��5���� 1028-1040.
����[47] Jacob A, Lancaster J, Buhler J, et al. Mercury BLASTP:Accelerating protein sequence alignment. ACM Transactions onReconfigurable Technology and Systems ��TRETS���� 2008, 1��2���� 9.
����[48] ����� ���ڸ�ͨ������ƽ̨��δ֪��ԭ������ϵͳ[D].������ �й������ž�����ҽѧ��ѧԺ�� 2016Li D C. Unknown Pathogen Detection System Based onHigh-throughput Sequencing Platform [D]. Beijing: Academy ofMilitary Medical Sciences, 2016.
����[49] Kurtz S, Phillippy A, Delcher A L, et al. Versatile and opensoftware for comparing large genomes. Genome Biology, 2004,5��2���� R12.
����[50] Zerbino D R, Birney E. Velvet: algorithms for de novo short readassembly using de Bruijn graphs. Genome Research, 2008, 18 ��5����821-829.
����[51] Li R, Li Y, Kristiansen K, et al. SOAP: short oligonucleotidealignment program. Bioinformatics, 2008, 24��5���� 713-714.
����[52] Quinlan A R, Hall I M. BEDTools: a flexible suite of utilities forcomparing genomic features. Bioinformatics, 2010, 26��6���� 841-842.
����[53] Huson D H, Mitra S, Ruscheweyh H J, et al. Integrative analysis ofenvironmental sequences using MEGAN4. Genome Research,2011, 21��9���� 1552-1560.
����[54] Darling A C, Mau B, Blattner F R, et al. Mauve: multiple alignmentof conserved genomic sequence with rearrangements. GenomeResearch, 2004, 14��7���� 1394-1403.
����[55] Thorvaldsdóttir H, Robinson J T, Mesirov J P. Integrative GenomicsViewer ��IGV���� high-performance genomics data visualization andexploration. Briefings in Bioinformatics, 2013, 14��2���� 178-192.
����[56] Krzywinski M, Schein J, Birol I, et al. Circos: an informationaesthetic for comparative genomics. Genome Research, 2009,19��9���� 1639-1645.
����[57]Ҷ��ǿ�����ܹܽ�ʯ���ߵ����Ͱ����ĺ�Ĭ֢С���ĺ������ѧ�о�[D].�������й������ž�����ҽѧ��ѧԺ�� 2016Ye F Q. Metagenomic Studies on The Biliary Microbiota ofPatients With Choledocholithiasis and The Gut Microbiota of MiceWith Alzheimer's Disease [D]. Beijing: Academy of MilitaryMedical Sciences, 2016.
����[58] Wang H, Chan S-H, Cheung J, et al. MICA: A fast short-readaligner that takes full advantage of Intel Many Integrated CoreArchitecture ��MIC���� arXiv preprint arXiv, 2014: 14024876.





