
诺氟沙星(NOR)、恩诺沙星(ENR)、环丙沙星(CIP)、达氟沙星(DAN)和沙拉沙星(SAR)为常见的氟喹诺酮类抗菌剂,目前在畜禽生产中被广泛应用. 此类药物使用不当导致的药物残留可能直接危害人体健康,也可能使致病菌产生耐药性,间接对人类健康造成影响. 多个国家和地区均规定了氟喹诺酮类药物的最高残留限量. 检测动物源食品中氟喹诺酮类药物残留的方法有微生物法、酶联免疫法(ELISA)、毛细管区带电泳法、电解分析法、高效液相色谱法(HPLC)、高效液相色谱 - 质谱联用法(HPLC-MS)等. 目前,在鸡肉产品的检测中,常采用的方法是《动物性食品中氟喹诺酮类药物残留检测 - 高效液相色谱法》(农业部 1025 号公告 14-2008). 本试验以此标准为基础,简化了试验前处理方法,优化了检测过程. 经试验验证,该法非常适合大批量处理样品,大量节省了处理时间和所需耗材,较大幅度降低成本,方法的灵敏度和回收率均可满足我国现行兽药残留检测分析的要求.
1 材料与方法
1. 1 药品和试剂
恩诺沙星对照品、环丙沙星对照品、诺氟沙星对照品、达氟沙星对照品、沙拉沙星对照品,均为中国兽医药品监察所产品,磷酸二氢钾、磷酸、三乙胺、氢氧化钠溶液(分析纯,广州试剂厂),乙腈(色谱纯,Fisher 公司).
1. 2 仪器和设备
Agilent 高效液相色谱仪( 美国 Agilent 公司) 配荧光检测器,高速匀质机及振荡器(德国 IKA 公司),高速台式冷冻离心机(美国 Thermo 公司).
1. 3 试液配制
分别称取恩诺沙星、环丙沙星、诺氟沙星对照品各10 mg,置于同一个 10 mL 容量瓶中,用 0.05 mol·L- 1的氢氧化钠溶液溶解定容至刻度,制得质量浓度均为 1 mg·mL- 1的混合储备溶液. 准确称取达氟沙星对照品10 mg,0. 05 mol·L- 1的氢氧化钠溶液溶解定容至 10 mL,使其质量浓度为 1 mg·mL- 1. 准确称取沙拉沙星对照品10 mg,0. 05 mol·L- 1的氢氧化钠溶液溶解定容至 10 mL,使其质量浓度为 1 mg·mL- 1.置 2 ~8 ℃冰箱中保存备用,有效期 1 个月.取上述恩诺沙星、环丙沙星和诺氟沙星的混和储备液 100 μL,达氟沙星储备液 20 μL,沙拉沙星储备液 200 μL,置于同一个 100 mL 容量瓶中,用流动相定容至刻度,配置成恩诺沙星、环丙沙星、诺氟沙星的质量浓度为 1 μg·mL- 1,达氟沙星质量浓度为0. 2 μg·mL- 1,沙拉沙星质量浓度为 2μg·mL- 1的混合标准溶液.提取液:称取 2 g 三氯乙酸于容量瓶中,加水至100 mL,制得的三氯乙酸溶液与乙腈按体积比 3 ∶ 1的比例混合,搅拌均匀,得到提取液.磷酸/三乙胺溶液:取磷酸 3. 4 mL,用水稀释至1 000 mL,配成 0. 02 mol·L- 1的磷酸溶液,用三乙胺调 pH 至 2. 4. 临用配制(易产生沉淀).
1. 4 测定方法
1. 4. 1 供试材料的制备 将市售的鸡肉样品用高速匀浆机绞碎得到供试试料,将空白鸡肉样品绞碎得到空白试料,在空白试料中添加适宜浓度的标准溶液作为空白添加试料.
1. 4. 2 提取与净化 称取 1 g 试料,置于匀浆杯中,加提取液 4 mL,高速匀浆 1 min. 匀浆液转入 15 mL聚丙烯离心管中,振荡 30 min,16 000 r/min,离心 8min,取上清液过 0. 2 μm 滤膜,装入进样瓶,待测.
1. 4. 3 色谱条件 色谱柱:Agilent TC-C18250 mm ×4. 6 mm(i. d),粒径 5μm. 流动相:V(0. 02 mol ·L- 1磷酸/三乙胺溶液) ∶ V (乙腈) = 80 ∶ 20. 流速:1mL·min- 1. 检测波长:激发波长 280 nm ;发射波长450 nm. 进样量:20 μL.
1. 5 线性范围与检出限
空白样品中加入混合标准液配制成的诺氟沙星、环丙沙星、恩诺沙星、达氟沙星、沙拉沙星系列标准溶液. 高效液相色谱仪检测后,以峰面积为纵坐标,药物质量浓度为横坐标,进行线性回归分析. 在空白样品中添加不同质量浓度混合标准溶液,按“1. 4”的方法进行处理,检测后取信噪比 S/N = 3 对应的样品浓度为方法检出限(LOD),信噪比 S/N =10 对应的样品浓度为方法定量限(LOQ).
1. 6 回收率和精密度
向空白待测样品中分别添加 3 个浓度水平的混合标准液,按“1. 4”的方法进行处理,每个浓度重复5 次,连续做 3 个批次,计算回收率和变异系数.
2 结果与分析
2. 1 色谱分析
在试验选定的条件下,测得诺氟沙星的保留时间为 6. 67 min,环丙沙星为 7. 19 min,达氟沙星为8. 18 min,恩诺沙星为 9. 07 min,沙拉沙星为 12. 93min,峰形尖锐,分离完全,空白组织样品在上述保留时间无干扰峰出现(图 1).
2. 2 方法的检出限及标准曲线
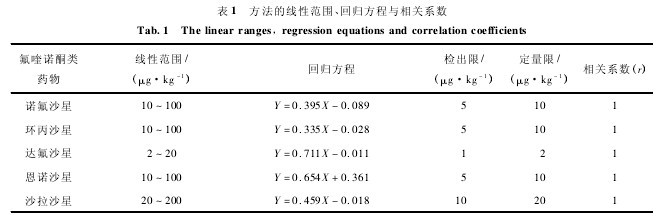
空白鸡肉样品中添加不同质量浓度的标准工作溶液 ,诺氟沙星、环丙沙星和恩诺沙星的残留定量限为 10 μg · kg- 1,达 氟 沙 星 的 残 留 定 量 限 为 2μg·kg- 1,沙拉沙星的残留定量限为 20μg·kg- 1.诺氟沙星、环丙沙星和恩诺沙星的线性范围为 10 ~100 μg·kg- 1,达氟沙星为 2 ~20μg·kg- 1,沙拉沙星为 20 ~200 μg·kg- 1. 对质量浓度不同的混合标准使用液进行色谱分析,工作曲线的线性范围、回归方程、相关系数见表 1. 可见,本方法灵敏度高、线性范围较宽,有一定的实用价值.【表1】
2. 3 方法的回收率与精密度
对鸡肉样品分别进行4 个浓度的添加试验,测得的回收率为84.71% ~99.66%,批内变异系数为0.20% ~4. 42% ,批间变异系数为 0. 33% ~2. 68% (表 2).
2. 4 抽样检测
从市场上购买 30 个鸡肉样品,每份样品 2 个重复,按本方法进行制样和检测,结果发现一例鸡肉样品中诺氟沙星残留量为 43. 5 μg·kg- 1,平行样品中诺氟沙星的质量浓度为 41. 4 μg·kg- 1,其余样品均未检出氟喹诺酮药物的残留. 表明该方法检测鸡肉中氟喹诺酮类药物残留量的重现性非常好,相对偏差保持在 3%之内,数据可靠.
3 讨论与结论
3. 1 样品前处理方法的优化
动物源食品中基质干扰复杂,富含脂肪和蛋白质,选择并优化样品的前处理方法对结果的准确性尤为重要. 本试验在农业部 1025 号公告-14-2008 所采用方法的基础上,对样品的前处理过程进行了一定的优化. 该标准检测方法采用磷酸盐缓冲液提取完后,离心取上清液,过固相萃取柱,从而除去样品中的杂质. 本试验的提取液由乙腈与质量分数为2% 的三氯乙酸按体积比 1∶ 3 混合而配得,蛋白质在酸性条件下变性,离心出沉淀除掉杂质,而提取液中加入乙腈也与流动相中乙腈比例吻合,出峰较好.比较其他相关研究,仲锋在鸡组织中检测氟喹诺酮类药物残留量的回收率为 70% ~107%,变异系数小于 10%;陈红等在鸡排泄物中检测氟喹诺酮类药物的回收率为 75% 以上,批内变异系数在2. 2% ~ 10% 之间,批间变异系数在 6. 2% ~ 13. 0%之间;而本试验测得的回收率为 84. 71% ~99. 66%,批内变异系数为 0. 20% ~ 4. 42%,批间变异系数为0. 33% ~ 2. 68% . 相较而言,本试验的回收率有了明显稳定的提高,精密度和准确度也相应改善了,定量限能满足日常采集样品的检测范围,而且检测的效率大大增加,节省了柱子的成本,减少了处理的时间,非常适用于样本量较大时的残留检测.
3. 2 检测方法的稳定性
将新鲜配制的混合标准工作液 50 μg·kg- 1于 4℃ 冰箱中避光保存 1、3、5、7 d 后上机检测,结果表明,被测药物保留时间基本不变,但浓度明显降低,可见标液在保存过程中不稳定,因此在测定过程中应该做到现用现配,处理完后的样品也应及时上机.
3. 3 结论
本试验在农业部 1025 号公告-14-2008 所采用方法的基础上,对样品的前处理过程进行了优化,使样品处理快捷便利,灵敏度高,重现性好,明显降低批量样品的检测成本,可有效应用于鸡肉食品品质的日常监控.
参考文献:
[1] 李俊锁,邱月明,王超. 兽药残留分析[M]. 上海:上海科学技术出版社,2002: 365-390.
[2] WINTER R W,KELLY J X,SMILKSTEIN M J. Antima-larial quinolones: Synthesis,potency,and mechanisticstudies[J]. Exp Parasitol,2008,118(4) :487-497.
[3] 朱蓓蕾. 动物性食品药物残留[M ]. 上海: 上海科学技术出版社,1994: 115.





