
����1 ����
������ѡ�����ܼ���,��Ƕ�ι�����Ũ�ȸ����ٽ罺��Ũ��ʱ,������װ�γ��ɿ����ԿǺͲ����Ժ���ɵĽ���,������װ����������ҪΪ��ͬǶ�μ�������ܼ�����.Ƕ�ι����コ��������Ϊҩ����������������,������ҽ�ò����Լ����������Ź㷺��Ӧ��.��Ƕ�ι����コ���ĺ˾��нᾧ��ʱ���dz�֮Ϊ�ᾧ�Խ���.���ڽᾧ����һ���������Է������γ���ά����ṹ�Ĺ���,��˽ᾧҲ�ɿ���һ������װ��������,ͬ�����ܼ�������һ��ͬ�����ᾧ��Ƕ�ι���������Һ�е�����װ��Ϊ[1].�ᾧ��ͨ���ϴ�,�ʽᾧ��һ�ֽ�ǿ��������,���������ڽᾧ�Խ���������װ����������������.��һ����,�ܼ��������¶ȡ�������Ũ�Ⱥ�Ƕ�γ��ȵȾ�������Ӱ��Ƕ�εĽᾧ��,�Ӷ�����ͨ���ı�ᾧ�������Ĵ�С,����ضԽᾧ��Ƕ�ι����������װ���̰����ᾧ�Խ�������̬�ͳߴ�Ƚ��е���.����,�ᾧ���̵��������羧��Ŀ�������Ҳ����Ϊ�ᾧ�Խ���������ͨ����Ƕ�ι����コ�������߱���һЩ�ص�.
��������Ƕ�ι������в��ܽᾧ��Ƕ�νṹ�IJ�ͬ,Ŀǰ�����¼���ᾧ��Ƕ�ι����コ�����о���Ϊ����.������Ҫ�����ں���ϩ��(��ҪΪ����ϩ(PE)�;۱�ϩ(PP))���ε�Ƕ�ι�����,���Winnik �ȶԺ��۶�ï������������( PFDMS) ��Ƕ�ι����������װ��Ϊ�����������о�.����,�Ժ���������ϩ(PEO)���ۼ�����(PCL)������������(PLLA)���۱�ϩ��(PAN)�;���ԵȽᾧ�����ε�Ƕ�ι����コ�����о�Ҳ�н϶౨��.���Ƕ�ι�����ᾧ�Խ������о���Ҫ�����ڽ�����ò�ͳߴ�ĵ����Լ��������������γɻ����ȷ���.���Ľ���Ӱ��Ƕ�ι�����ᾧ�Խ�����ò����Ҫ���ء��ᾧ�Խ����Ļ��������Լ�"Ƕ�ι�����"���Ʊ���������.
����2 Ƕ�ι�����ᾧ�Խ�������ò����Ӱ������
����Ƕ�ι���������Һ���������γ������ܽϵ͵Ľ�����̬,������������Ϊ�����Dz�������(��Ҫ��ӳ�������ε��ų�̶�)���˿ǽ���������(�ɲ�ͬ�������ܼ���ı��������ͽ��������)�ͽ����˵�������(��ӳ�������ε���չ�̶�)֮��[2 ~5].
�������ڽᾧ�Խ���,Halperin ����Ϊ�˵Ľᾧ������ռ������λ[6],��˽����������γ�Ƭ��ṹ;��Ƭ��ṹ�ֵ��¿Dz�Ƕ�ν��ܶѻ�,���¿������μ���ų�������,����Dz������ܵ�����,�ʸ����ʵĽ��漴���״���������ڿDz������ܵĽ���.�����������صľ��������˽ᾧ�Խ�������ò.����������ṹ���ܼ����ʡ��¶ȵ����س���Ӱ��Ƕ�ι����ﲻͬ���ε������ܼ�������,ҲӰ��ᾧ���εĽᾧ�Ժ�������,�Ӷ�Ӱ��ᾧ�Խ�������̬.
��������,�ᾧ�Խ�������̬Ҳ������Ƕ�ι�������ܼ��еĽᾧ;��(�Ʊ�����).
����2. 1 ���������ṹ��Ӱ��
��������������ṹ�ǽᾧ�Խ�������װ��Ϊ������,����Ƕ�γ��ȡ�Ƕ�α�����Ƕ�νṹ�ȶ���Ӱ��ᾧ�Խ�������ò[7,8].����[9]�о��˾ۼ�����-b-��������ϩ(PCL-b-PEO)��ˮ��Һ�еĽ�����̬,�������Žᾧ���� PCL ���ȵ�����,������̬������(��ͼ 1A ��ʾ)��Ϊ�̰�״�����״(��ͼ1B ��ʾ) ��Ƭ״( ��ͼ 1C ��ʾ) .�������ù�һ����֦�ܶ����۶�������˰붨���Ľ���.�ڽᾧ�Խ�����,�������οɿ����ǽ�֦�ڲ������εľ������.��һ����֦�ܶ�(��σ)�Ķ���Ϊ����������������������״̬����ռ�����(πRg2,Rg2Ϊ������ת�뾶)�����ھ��������ռ���(S)֮��.���� Rg2��������εĹ����й�,�ܿ������εij��ȡ��ܼ����ʺ����Ӽ���Ӱ��,�� S ��ᾧ���εij��Ⱥ����ھ����е��۵������������ṹ�й�[9].��σ ��ֵԽ��,������������֮��Խӵ��,������������ξ���IJ���渲��,���¾������������.���Ƿ��ֵ���һ����֦�ܶ� ��σС��һ���ٽ�ֵʱ(ԼΪ 3. 0 ~ 4. 8),���������γ�Ƭ״��ò;���� ��σ ����,���������γ���״(��ͼ 2 ��ʾ)[9].���ڲ�ͬ�����Ƕ�ι�����,��ᾧ�Խ�����̬��Ƕ�ι�����ṹ�仯�����������Ƶ�[10 ~ 12],���������н�����̬���ܹ۲쵽,�纬�ᾧ�Ծ�ϩ�����ε�Ƕ�ι�����Ҳ�����γ��������ε���״��Ƭ״�ᾧ�Խ���[13 ~16].�ھ۶�ï������������-b-�������ϩ(PFDMS-b-PI)Ƕ�ι�������,���Žᾧ���� PFDMS������,������̬Ҳ���ɰ�״��ΪƬ״[17].Mihut��ͨ�����ھ۶���ϩ-b-��������ϩ(PB-b-PEO)Ƕ�γ��Ⱥ�Ƕ�α���,Ҳ�õ�����״����״�����״�Ȳ�ͬ�Ľ�����̬[18].Hillmyer ��[19]���ֵ��۶�����ϩ����-b-����ϩ(PDMA-b-PE)�п���Ƕ�� PDMA �ı����ﵽ30 wt% ����ʱ�γɱ�����״����( ��ͼ 1D ��ʾ),���� PDMA Ƕ�γ��ȼ�С,���״����������״����(��ͼ 1E ��ʾ)��֮����.����,Ҳ�����ױ�����һЩ��̫�����Ľᾧ�Խ�����̬,�� Zhu ���ھ���ϩ-b-��������ϩ(PE-b-PEO)Ƕ�ι������ˮ��Һ�й۲쵽��˵Ľᾧ�Խ�����̬(��ͼ 1F ��ʾ)[20],�� PCL-b-PEO ��ˮ��Һ�з��� PCL �Ľᾧ���յ������˵Ķ��Ƭ���ṹ[21],�ڵȹ�۱���ϩ-b-��������ϩ(iPS-b-PEO)��ˮ��Һ�й۲쵽��״�������γ�(��ͼ 1G ��ʾ)[22].Wang �ȷ���֧���۶�������ͪ(���ܵĽᾧ����) /��������ϩ(PPDOstar-b-PEG)Ƕ�ι�������ˮ�п��γ�����״�Ľ���(��ͼ 1H ��ʾ)[23,24].

����Ƕ�ι���������ṹ����Ӱ��ᾧ�Խ�������̬,Ҳ��Ӱ����ͬ��̬�ᾧ�Խ����ijߴ�.������״����������-b-�۱�ϩ��(PLLA-b-PAA) ������˵,O'Reilly ��[25]������״�����ij�����ᾧ��Ƕ�� PLLA ���ȵ����Ӷ���С;��������Ƕ�� PAA ���ȵ�����,���½����Dz�������.��һ����,����Ƕ�ε��ܽ��Ծ����˵������ܽ��,�Ӷ�Ӱ���˽ᾧ�����ijɺ����ʺ���������,�Dz�Ƕ�ε����ܼ���Խ��,��״�����ij���Խ��[26].
��������Ƕ�γ��Ⱥͱ���,�ᾧǶ�εĽᾧ���ԺͿ������ε���˳��Ҳ��Ӱ��ᾧ�Խ�������̬.�۶�ï�������һ�����-b-�������ϩ(PFDES-b-PI)�� PFDMS-b-PI �ṹ����,�� PFDES-b-PI ����������װΪ�ͽ������ʵĽṹ[27],���������߹�����PFDES �ϸߵ���������ᾧ��. PFDMS-b-PBLG(PBLG Ϊ�۹Ȱ�������)�� PFDMS Ƕ����� PBLGǶ�νϳ�������ʱ�����γ�Ƭ״����,������Ƕ�α������Ƶ������� PFS ��Ƕ�ι�����ȴ�����γ�Ƭ״�ṹ.�������� PBLG Ƕ�εĸ��Խϴ�,�� N,N-����������( DMF) �в�ȡ�������Ե�����������Ǹ���ͬ�Ե������Ź���,���¿Dz�λ�轵��,�Ӷ��γ�Ƭ״����[28].�ڹ����������������ܼ��Ҳ��ᾧ��Ƕ��Ҳ���ƻ��ᾧǶ�εĽᾧ,�Ӷ����ؽ�����ò.�ھ۶���������(PDMS)��ѡ������ �� ��,�� Ƕ �� �� �� �� PFP-b-PFDMS-b-PDMS(PFP Ϊ�۶�ï����������) �е� PFDMS Ƕ�ο��Խᾧ,PFP Ƕ�β��ܽᾧ.�� PFP Ƕ�ξۺ϶ȵ�ʱ,���Եõ�����״����;�� PFP Ƕ�ξۺ϶ȸ�ʱ,��ֻ�γ���״����[29].������������ PFP Ƕ�γ��ȵ�����,��� PFDMS Ƕ�νᾧ���ƻ�����Ҳ����,���յ��� PFDMS Ƕ�����ᾧ,�γ���״����.
������������Ƕ�ι�������ѡ�����ܼ���Ҳ������װ�γɸ�����̬�Ľ���,Ϊ��ȷ�����ַ����νᾧ�Խ������γ����ɽᾧ������,����ͨ���������ַ���������֤:(1)�������νᾧ�Լ������ᾧ���ε��۵�����,���ֽ�����̬��Ϊ����[30].ͬʱҲ���ֽᾧ���յ����ε����ν���ת��Ϊ��״����[31].(2) ���ýṹ�ͳ������Ƶ�������������ᾧ������,���۲쵽���ν���[13,32].����������˵��,�ᾧ��Ƕ�ι���������νᾧ�Խ������γɹ�����������������.��һ����,��ʹ�ᾧ�Խ��������ν���������ͬ����̬,���ߵijߴ硢�ٽ罺��Ũ�ȡ�����Ҳ���ܲ�ͬ[33,34].
����2. 2 �ܼ����ʵ�Ӱ��
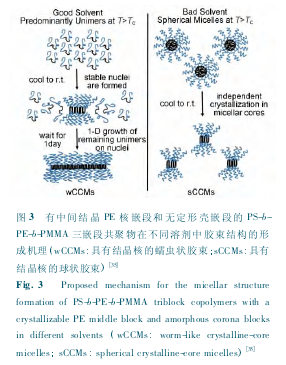
����Schmalz ���о��˾۱���ϩ-b-���� ϩ-b-�ۼ���ϩ�����(PS-b-PE-b-PMMA)��Ƕ�ι������ڲ�ͬ�ܼ��е�����װ��Ϊ[30,35].���ڸ����¼ױ��� PE ���нϺõ��ܽ���,����ʱǶ�ι������γ���ȫ�ܽ�ĵ�����,����Һ����һ���¶Ⱥ� PE ���η����ᾧ,��ֻ�γ���Ŀ���ٵĽᾧ�Խ���,�������������ڽᾧ�Խ����ľ��������������,�Ӷ��γ����״����.���ڶ�?���м��ȵ�PE ���۵�����ʱ���� PE ���ܽ��Խϲ���Ƕ�ι������γ����ε����ν���,���¶Ƚ���ʱ PE �������ν����з������ᾧ,�����������β���(��ͼ 3 ��ʾ)[35].Han ��[36]�о����ܼ�ѡ���Զ� PS-b-PLLA Ƕ�ι����コ����̬��Ӱ��.���Ƿ����ڶ� PS ����ѡ���Ե��ܼ����� PS-b-PLLA �γ���״����,������ PS Ƕ�εĸ�ѡ�����ܼ�����̼(CS2)��,���� CS2����������,������ò�����ɶ̰���Ƭ״��ת��.������Ϊ�ᾧ�Ժ˲�������ܼ�֮����������Ǿ���������̬����Ҫ����,�� Halperin ���������[6].���ǻ��� PS-b-P2VP-b-PEO(P2VP Ϊ�� 2-��ϩ�����)�ļױ���Һ�м�������ˮ,�����ڽ��������½�����̬��Ƭ״��Ϊ����[37].PFDMS-b-P2VP Ƕ�ι�����Ľ�����̬���ܼ����ʵ�Ӱ��Ҳ�dz�����,�ڼ״��ܼ���Ϊ��״,����������ܼ���Ϊ��״,������Ϊ����������� PFDMS �Ľᾧ[38].
��������ͬ����ԭ��,������ PI250-b-PFDMS50��ѡ�����ܼ��м����������ܼ�����������߰�״�ᾧ�Խ����ij���[39].Park �ȷ��� POT-b-PEO(POT:���������)��ˮ���γ����ν���,���ڼ״����γ���ά״����[40].Su �ȷ��� P2VP199-b-PCL310�� DMF/ˮ����ܼ����γ�Ƭ״��������,���ڶ� PCL �����ܽ��Խϴ������� ( THF) �����γ��� �ν���[41].�ܼ��Խᾧ�Խ����˵ĽṹҲ���ܲ���Ӱ��.Zhu ��[42]���� PE-b-PEO �ڹ��ܼ� 1,2,4-���ȱ��и����·�ɢΪ����,���º������м�Ϊ�������ϩƬ������Ϊ���� PEO �����״����;����ѡ�����ܼ� DMF �и������γ���״����,���º�ת��Ϊ�ڲ�˫��Ƭ������״����.
������������Կ���,�ܼ���Ƕ�ι�����ᾧ�Խ�����Ӱ��Ҫ�ȶ����ν�����Ӱ����Ӹ���,������Ӱ��������εĹ�����ܽ���,ҲӰ��ᾧ���εĽᾧ�ԡ��ᾧ���̺ͽᾧ��̬.����,���Ρ�pHֵ��Ҳ�����Ÿı佺�����ܼ�����,�Ӷ��յ��ᾧ�Խ�������̬�����ı�.���Ƿ������ε�"����"ЧӦ���Խ��� PEO Ƕ����ˮ�е��ܽ���,ʹ�����ųߴ��С,���½����Dz����εĹ�һ����֦�ܶ� ��σ����,PCL-b-PEO ����������-��ת�������-Ƭת��[43].��ͬ�����յ� PCL-b-PEO �ᾧ�Խ���������̬ת���������ͬ,�����������ӵ�"����"ЧӦǿ�ȼ���������й�[44].��Һ pH ֵҲ��PCL-b-PEO ˮ��Һ�еĽ�����̬��Ӱ��.PCL66-b-PEO44�����Ժ�����ˮ��Һ���γ���״����,����ļ��������������-��ת��.���������ڼ��Ի����� PEO Ƕ����ˮ���������ñ��ƻ�,ͬ������Dz����ι�һ����֦�ܶ� ��σ�Ľ���[45].
����2. 3 �ᾧ�¶� / ����
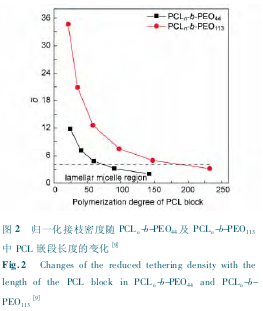
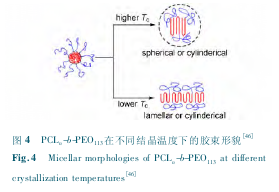
�������ǿ����� PCLn-b-PEO44�� PCLn-b-PEO113����ϵ�еĹ����������װ,���ֽᾧ�¶ȶԽ�����̬����˫������[46].��߽ᾧ�¶ȿ��Լ��� PCL���ε��۵�����,���� PCL �ľ�Ƭ���,�Ӷ����½�֦�ܶ�����,�����γ���״����;����һ����,��߽ᾧ�¶Ȼ�ʹ�����˵Ľᾧ��������,�������������������ϵ�е��������ø�������,�Ӷ������γ�Ƭ״����.���� PEO ���νϳ��� PCLn-b-PEO113Ƕ�ι�������˵,��һ�����ø�������(��ͼ4 ��ʾ) ;������ PEO ���ν϶̵� PCLn-b-PEO44Ƕ�ι�����,�ڶ������ø�������.
����Zhu �ȵ��о������¶ȿ������� PE-b-PEO ��ˮ��Һ�н�����ò�����ű仯.������ PE-b-PEO��ˮ�п����γɺ��ж������ϩ�ᾧ�ں˵���״����,���¶����߿ɵ��¶������ϩ�ᾧ�ں˷����ں�,ת��Ϊ������״����.Manners �� Winnik �ȶԺ� PFDMS �ĽᾧǶ�ι����������ϵͳ�о�.���Ƿ����¶ȿ�������PFDMS40-b-PDMS480�����İ�״-��״�Ŀ���ת��.
�����пչ�״�ṹ���γɿ����� PFDMS �˱���������PDMS Ƕ�����ռ������ͬ���õĽ��.�������¶�����,������� PDMS ���ܽ��Ա��,�Dz�Ƕ�����͵��½�����ȡ��״�ṹ[47].PFDMS-b-PBLG�����������γ�ĩ����ά״�ıⳤ�ν���,���ڽϸ��¶��ȴ���������״ת��Ϊ���ӹ����һԲ������Բ��Ƭ״����.����������òת���ԭ���������ȴ���ʹ�����������Գɺ˹���,�������ܽⲢ�ᾧ�����ھ��˱���,ʹ����ò���Ӿ�һ[28].

����Mihut ���о��� PB-b-PEO �� PB ��ѡ�����ܼ��������е�����װ��Ϊ.����Һ�� PEO �۵�����(��ʱ�γ��������ν���)���µ� -30 ��ʱ,PEO����״�������ڽᾧ;Ȼ�����͵����͵�Һ���¶�ʱ,�γ��˽ᾧ��״����(��ͼ 5 ��ʾ)[48].�������ڵ������������ PB Ƕ�ε��ܽ������ϲ�,PB Ƕ�δ�������״̬ռ�ݿDz�ռ����.��һ����,������Һ����ȴ�� 30 ��,��Ϊ PEO �Ͽ�ijɺ������γ�Ϫ��״�ۼ���[49],���� 30 ������,��Ϊ���������������γ�Ť����Ƭ״�ṹ[50].�ɴ˿ɼ��¶ȼȶԿ��������ܽ�����Ӱ��,Ҳ�Խᾧ���εĽᾧ�Բ���Ӱ��,���߾���Ӱ�쵽�ᾧ�Խ�������̬.
����3 Ƕ�ι�����ᾧ�Խ����Ļ�������
��������ͨ�����п�����������,��������Ƕ�ι�����ᾧ�Խ����б���������,���ø����Կɵ��ؽᾧ�Խ����ijߴ硢��״����ʵ�ֲ�ͬ�ᾧ�Խ���֮�������[51],��Խᾧ�Խ����Ľṹ-���ܹ�ϵ�о�����Ӧ������Ҫ����,��ΪǶ�ι����コ��������ͨ��������״�ͳߴ��������[52].�����Ƕ�ι���������ν���,���������ǽᾧ�Խ��������е�����.�Ƚϳ����Ľᾧ�Խ�����������Ϊ�ܽ�ĵ������ڽᾧ�Խ����Ͻ��и�������,�û���Ҫ�������ܼ��Խᾧ������Ҳ����һ�����ܽ���,ʹ����ϵ�д��ڽ϶���ȫ�ܽ��Ƕ�ι����ﵥ������,ͬʱ�����������ᾧ�Խ�����Ϊ��������������,��������ʽ�ɲ������ַ�����ʵ��.��һ�ַ���������ڽᾧ�Խ������ӵ���Һ�м���һ�����ܽ��ڹ��ܼ��е�Ƕ�ι�������Һ.Winnik �ȷ����� PI550-b-PFDMS50�̰�״�ᾧ�Խ����ļ�����Һ�м��벻ͬ�� PFDMS-b-PDMS �� THF ��Һʱ,�ڰ�״���������˻ᷢ������,�䳤������빲���ﵥ���;��ֵ�������(munimer/ mseeds)�����Թ�ϵ(��ͼ6 ��ʾ)[53],��֤���ᾧ�Խ������л�������������.�� �� �� �� �� �� �� �� ʵ �� �� �� �� �� �� �� ��PFDMS ���ε�Ƕ�ι����Լ����� P3HT ��Ƕ�ι�����Ļ�������[54 ~ 56].����Ҫע�����,���ܼ��ļ�����Ӧ������һ����Χ,�����Խᾧ�Խ�����������������Ӱ��,��ʹ�������ȱ��,�ֲ����[57].�ڶ��ַ�����������߷��ӵ���ʱ���õ�"�Ծ���"����,�����ᾧ�Խ�����Һ���ȵ�һ���¶Ȳ�����һ��ʱ��ʹ�ֽ����ܽ���������ٲ��ֽᾧ�Խ�����Ϊ����,Ȼ����Һ�������ʵĽᾧ�¶����ܽ��Ƕ�ι����ﵥ���ھ�����������.
��������һ�ַ�����,�����¶ȡ�ʱ����ܼ����ʶԲ������ֵ���Ŀ����ҪӰ��[58],���ᾧ�¶�����ҪӰ���������ʺͽ�����̬[59].����"�Ծ���"����,Winnik ��ͨ���ı侧����Ŀʵ���� �� �� �� ��PFDMS ���ε�Ƕ�ι������״�ᾧ�Խ������ȵĵ���[60 ~ 62].Dove ��Ҳ���ø÷����� PLLA-b-PAAʵ���˻�������,�۲쵽��״�ᾧ�Խ����ij�����ᾧʱ������Թ�ϵ[63].

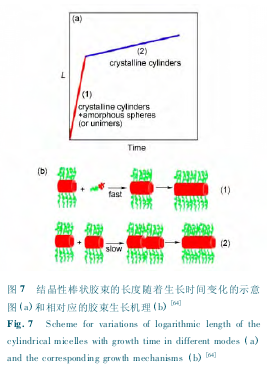
�����ᾧ�Խ�������һ��������ʽΪ��ͬ����֮���ż��.���Dz�������"�Ծ���"�ķ��������Լ�ͼ 7 �ᾧ��״�����ij�����������ʱ��仯��ʾ��ͼ(a)�����Ӧ�Ľ�����������(b)[64]Fig. 7 Scheme for variations of logarithmic length of thecylindrical micelles with growth time in different modes(a)and the corresponding growth mechanisms ( b)[64]���л��ܼ��ķ����о��� PCL-b-PEO �ᾧ�Խ�����ˮ��Һ�е���������ѧ,���ְ�״�ᾧ�Խ����ij�����ʱ��ı仯�ɷ�Ϊ������,��һ���ε������ٶȽϿ�,������Ϊ���Ӧ���ܽ�ĵ��������ھ��ֽ����ϵ�����,�ڶ����ν�����ʱ��仯�����ʽ���,�������ڲ�ͬ��״����ͨ��β��֮���ż������(��ͼ 7 ��ʾ)[64].Winnik ���Ʊ����ɶ��������ɵ� PFDMS-b-P2VP Ƭ״����[65],���õ�������֤���˽����ڲ��Ķྦྷ�ṹ,����״�����ڲ�Ϊ�����ṹ[66],��ܺõ�֧���˽ᾧ�Խ�����ż��-��������.һЩ������״��Ƕ�ι�����ᾧ�Խ� ��,�� �� ״ �� iPS-b-PEO �� �� �� �� �� ״ ��PPDOstar-b-PEG ����������Ϊ��Ƭ״�ᾧ�Խ������϶��γɵ�.
����4 Ƕ�ι�����
��������Һ���ܽ��Ƕ�ι��������γɾ��ֵ�Ƕ�ι�������в�ͬ�Ľṹʱ,�������п��ܽ��и�������,��ʱ�γ�"Ƕ�ι�����"."Ƕ�ι�����"�ǽ�����ոճ��ֵ�һ������,����ζ���Խᾧ�Խ���Ϊ������Ԫ,ͨ����������װ�γɸ��߲�εĽṹ.
����Winnik ��[67]��һ���Ʊ����м�Ϊ PFDMS53-b-PI320����������Ϊ PFDMS48-b-PMVS300(PMVS Ϊ�ۼ���ϩ��������)�����İ�״��Ƕ�νᾧ�Թ�����(��ͼ 8A ��ʾ).Ȼ�����ǽ� PI �Dz㽻������빲�ܼ�,ʹ���������˵� PFS-b-PDMS �ܽ�,��λ���в��������� PFS53-b-PI320����������.��˵���ᾧ�Խ���������Ϊ����������"Ƕ�ι�����",���Ұ�״�������ij���Ҳ���������ں�����Ƕ�ι��������,��˿���ͨ������Ƕ�ι��������������ȷ���ع������Ľṹ.ͨ�����ֶಽ�����ķ���,���ǻ������˻��� PFDMS Ƕ�ι��������Ƕ��[68](��ͼ 8B ��ʾ )�Ͷ�Ƕ��[69]�Ĺ�����.����,���ǻ���ӫ��Ƕ�ι����コ����װ��һ��,ͨ������ÿ�ν����ķ������ʵ����Ƕ�ι������������ɼ�����Χ�ڵ���ɫ����[70].��Ҫָ������,�����Ĺ�������������ܻ���ݽ����Ľ�ɫ,���½ᾧ����״����ĩ��ż������.�������ͨ�������ڼ���Ĺ������Ƕ����Խ϶�,�����ڼ�����Ӧ�ɺ�Ƕ�εľ�����ʱ����[71].
�����������������ɷ����ڰ�״����������,�����Է�����Ƭ��״�Լ���Բ״�����ı�Ե�Ӷ��γ�"Χ��״"������[68](��ͼ 8C ��ʾ)������������Բ״������[72](��ͼ 8D ��ʾ).Winnik �������� PFDMS ������������������������ PFDMSǶ�ι����コ��,�ɹ��Ʊ��˶�����ν���,�Լ����ι�����[73],�ұ۵ij��ȿɿ�.��������ֳɹ��ؽ��˽�ϸ�İ�״�������������ں˽ϴֵİ�״�������ֵĻ���ĩ��,�Ӷ��Ʊ�����֦״������[74].���Ƿ��ֽ�ϸ�Ľ��������ھ��ֻ��Ա������ʼλ�õľ��������˽������յ�֧���̶�,��Ϊ���ֵ��м�ν����ij��Ⱥͽ�֦��״�����ij��Ⱦ�����PFDMS Ƕ�εĻ�������������,�������һ�ֺܺõĿɿ��Ʊ���֦״�����ķ���.��ͬ�ṹ��Ƕ�ι������γɽᾧ��"Ƕ�ι�����"��һ�������Ǿ��ֽ����еijɺ�Ƕ������Ƕ�ι������нᾧ���εľ�����ƥ��.���ʵ����ʾ PFDES-b-PI ��������Ϊ����ƥ�䲻�������� PFDMS-b-PI ��״������ĩ��[27],�� PFDMG-b-PI ��Ϊ����ƥ�����ʵ������[68].��� Winnik �� Manners ��[75]�ϳ���һ����Ƕ����Ԫ������ PI-b-(PFDMS-grad-PFDES),�ݶȹ��۵ĺ�Ƕ�α�������Ӧ PFDMS �� PFDES �Ľᾧ��,���ǽ����Ʋ����� PFDMS �ľ�����Һ���ȼ�����������Ƕ����Ԫ�����ﵥ����Ϊճ���,�ټ�����һ�� coil-b-PFDES ����,Ҳ�����Կ˷����ֽᾧ�˵ľ���ƥ���Լ���ͬ������װ����ѧ,ʵ�ֺ�PFDMS �� �� PFDES Ƕ �� �� �� �� �� ��"Ƕ �� ������".
����"Ƕ�ι�����"���γɲ��������ں� PFDMS ��Ƕ�ι�����,�����������������Ҳ�ɹ��Ʊ�"Ƕ�ι� �� �� ". Schmalz �� ʹ �� PS-b-PE-b-PMMA(SEM)�� PS-b-PE-b-PS (SES)������Ƕ�ι�����ɹ��Ʊ��˿ɿص���Ƕ�ι�����,�����ڿDz���Թ۲쵽�������ṹ(��ͼ 9 ��ʾ)[76].Winnik ��Ҳ�Ʊ��˻��ھ����������Ƕ�ι��������Ƕ�ι�����[77].

����Ƕ�ι�����ᾧ��"������"Ҳ�ɽ�һ������װ�γɽṹ���Ӹ��ӵ�"��������".Winnik �ȷ��� PFDMS-b-P2VP/PFDMS-b-PDMS/PFDMS-b-P2VP�γɵ� A-B-A ��Ƕ��"������"���м�ν��� B �ܹ�������γɲ�ͬ�ṹ��"��������",����"��������"�Ľṹ��ͨ�� A �� B �̽����ij���������[78].

����ͨ�������,��������������������ͬʱ���е�,�õ��Ľᾧ��"Ƕ�ι�����"�������ĶԳ���,�������ĶԳ�"Ƕ�ι�����"���Ʊ�����һ�������ս.�����ĶԳƵĹ���������ڴ�ͳ���������и���ļ���,������������װ��Ϊ"��������".

����Winnik �Ƚ�ϸ���������ѡ���Խ��������Dz�ķ����ɹ����Ʊ����������� A-B �� A-B-C ��"Ƕ�ι�����"[79].���������� PFDMS-b-PDMS ��״�������������� PFDMS-b-PI �γ� ABA ������,���Ž� PI Ƕ�ν���(��ͼ 10A ��ʾ),���� PFDMS-b-PI�ν������پ�����������,Ȼ���ڼױ����ܽ�PFDMS,�õ����˾����������Ե� PFDMS-b-PI ����,֮���ټ��� PFDMS-b-PDMS �ɹ��õ��˵��������� PFDMS-b-PDMS/PFDMS-b-PI A-B ��"��Ƕ�ι�����"(��ͼ 10B ��ʾ).������������һ��Ƕ�ι�������Һ�ɵõ� A-B-C ��"��Ƕ�ι�����"[79].
����5 ���ۺ�չ��
������������Կ���,���ڽᾧ�������������ܼ����õ������������ľ���,���ǿ��Ը��ӷ���ص���Ƕ�ι�����ᾧ�Ե���ò.ͬʱ,�������ν���,�ᾧ�Խ����������γɸ߶ȸ������Ե���ò,����ӵ�пɿ������Լ��γ�"������"�Ķ�������.
����Ŀǰ�Խᾧ�Խ������о��dz���Ծ.Winnik���Ѿ��ܹ��ں� PFDMS �����л����ε�Ƕ�ι������жԽ����ijߴ����״���нϺõؿ���,���ɹ��Ʊ��˶�����״��Ƕ�ι�����,ͬʱ����˵��������ڽ����ᾧ�Ժ��������Ļ���.���ڽᾧ�Խ������о�Ҳ�Ѿ���չ���˴��������Ĵ��л�Ƕ�ι�������ϵ,������Ҫ�����ڽ�����̬���о�,��������Щ��ϵ�������������ߴ���ƺ�Ƕ��"������"���о��д�����.������Ϊ,��������о���������������������Ҫ��һ��Ŭ��:(1) Ƕ�ι������ر��Ǵ��л�Ƕ�ι�����ᾧ�Խ�������̬���ƺ������������д��������о�.�粻ͬ�ᾧ�Խ������ż������[80],�������кܶ�ʵ��֤��֧������������ʽ�Ĵ���,�������������в���ȷ.���ڴ��л�Ƕ�ι�����Ĺ㷺��,̽����Щ��ϵ�нᾧ�Խ�������������̬���ƺ��������������ڽ�һ���ƶ��ᾧ�Խ�����Ӧ��.(2) Ƕ�ι�����ᾧ�Խ���������ͨ����һ��������ĵ������,ʵ�ֽᾧ�Խ����Ŀ�����������������װ���ƺͽᾧ�����۷��滹������Ӧ�÷��涼������Ҫ������,��Ŀǰ��δ���йظ÷���ı���.(3) ��ν���ͬ���ܼ����ڽᾧ��"������"��,ʵ���ɽṹ�����ܵ���װ������δ����������о�����֮һ.Ŀǰ���ں�PFDMS �Ľᾧ��"������"�г���ʵ��ijһ����Ƕ�ε�ѡ���Թ��ܻ�[81 ~ 83],���йز�ͬ���ܵ���װ�Լ�������ϵ��Ƕ��"������"���Ʊ������ٱ���.
������ �� �� ��
����[1] He W N,Xu J T. Prog. Polym. Sci. ,2012,37: 1350.
����[2] Yu Y S,Eisenberg A. J. Am. Chem. Soc. ,1997,119: 8383.
����[3] Zhang L F,Eisenberg A. Polym. Adv. Technol. ,1998,9:677.
����[4] Zhulina E B,Adam M,LaRue I,Sheiko S S,Rubinstein M.Macromolecules,2005,38: 5330.
����[5] Larue I,Adam M,Zhulina E B,Rubinstein M,Pitsikalis M,Hadjichristidis N,Ivanov D A,Gearba R I,Anokhin D V,Sheiko S S. Macromolecules,2008,41: 6555.
����[6] Vilgis T,Halperin A. Macromolecules,1991,24: 2090.
����[7] Wang J,Zhu W,Peng B,Chen Y M. Polymer,2013,54:6760.
����[8]Makino A,Hara E,Hara I,Ozeki E,Kimura S. Langmuir,2014,30:669.
����[9]Du Z X,Xu J T,Fan Z Q. Macromolecules,2007,40:7633.
����[10]Xu J T,Fairclough J P A,Mai S M,Ryan A J. J. Mater.Chem.,2003,13:2740.
����[11]Xu J T,Jin W,Liang G D,Fan Z Q. Polymer,2005,46:1709.
����[12]Kamps A C,Fryd M,Park S J. ACS Nano,2012,6:2844.
����[13]Yin L G,Hillmyer M A. Macromolecules,2011,44:3021.
����[14]Wang W,Liu R,Li Z,Meng C,Wu Q,Zhu F. Macromol.Chem. Phys.,2010,211:1452.
����[15]Zhao Y,Shi X B,Gao H Y,Zhang L,Zhu F M,Wu Q. J.Mater. Chem.,2012,22:5737.
����[16]Liu R,Li Z Y,Mai B Y,Wu Q,Liang G D,Gao H Y,Zhu FM. J. Polym. Res.,2013,20.
����[17]Cao L,Manners I,Winnik M A. Macromolecules,2002,35:8258.
����[18]Mihut A M,Crassous J J,Schmalz H,Drechsler M,Ballauff M.Soft Matter,2012,8:3163.





