
microRNAs(miRNAs)是一类长度为18~22nt的单链小RNA,位于基因组的非编码区,具有高度保守性、时序性和组织特异性等特点。成熟的miRNAs通过RNA沉默复合体(RISC)特异性识别靶基因mRNA的3′非翻译区(3′UTR),在转录后水平调控基因表达,进而引起靶基因的翻译抑制或mRNA降解,对基因表达起负调节作用。
目前,在小鼠中已经发现1908个成熟的miRNAs,其中,部分miRNAs已证实参与调节生物体生长、发育和疾病发生过程中相关基因的表达。对miRNAs功能深入研究,有利于对生物体生理、病理机制的理解,并为疾病诊断和治疗以及动物育种提供理论基础。
小鼠局部粘着斑激酶(focal adhesion kinase,FAK)基因的第22个内含子转录后经一系列剪切形成miR-151。研究表明,miR-151在多种癌症,如膀胱癌、肺癌、前列腺癌、乳房癌、直肠癌和肝癌等中表达量呈增高趋势,并参与癌细胞增殖和病灶转移等病理过程,对肿瘤形成有重要影响。
miR-151包括2个成熟的miRNAs:miR-151-5p和miR-151-3p,两者均来自miR-151的初级转录本,但含有不同的种子序列,能够靶向不同的基因。
miR-151-5p与其宿主基因FAK共表达,影响癌细胞的转移,并能通过靶向抑制因子RhoGDIA,激活下游肿瘤发生相关因子Rac1、Cdc42和RhoGTPases,引起肝癌细胞恶性转型和侵袭转移;miR-151-5p在人前列腺癌细胞(PCα)中高表达,通过抑制癌细胞生长的木黄酮处理后,miR-151-5p的表达受显著抑制,计算机模拟和双荧光素酶报告分析证明,肿瘤抑制因子Casz1、IL1rapl1、Sox17、N4bp1和Arhgdia均是miR-151-5p的靶基因,miR-151-5p通过靶向这些抑癌因子,引起癌细胞扩散。当前,miR-151-5p在肿瘤细胞上的功能已部分阐释,而对来自同一个初级转录本的miR-151-3p功能研究报道还很少,因此,对miR-151-3p表达调控的研究有利于深入了解其在不同组织中发挥的功能。
本研究通过对miR-151-3p保守性分析和靶基因预测,并利用实时定量和双荧光素酶分析系统对靶基因进行筛选和鉴定,旨在寻找miR-151-3p靶位点中与小鼠肌肉发育和肿瘤发生相关的基因,为进一步研究miR-151-3p在生物机体中的功能奠定基础。
1、材料与方法
1.1试验材料
1.1.1质粒和细胞系小鼠成肌细胞系C2C12购自美国ATCC,DH5αCompetentCells购自北京康为世纪生物科技有限公司,pMD19-TVector购自日本Takara公司,HEK293T细胞、psi-CHECKTM-2质粒载体由西北农林科技大学动物科技学院动物脂肪沉积与肌肉发育实验室保存。
1.1.2试剂和仪器DMEM高糖培养基、胎牛血清均购自美国HyClone公司,马血清购自美国Gibco公司,胰蛋白酶购自美国MP公司,Hepes购自上海生工生物工程技术服务有限公司,RNAisoPlus、PrimeScript RRTreagentKit、SYBR RPremixExTaqTMⅡ、XhoⅠ、NotⅠ和T4DNALigase均购自日本Takara公司,Tissue Di-rect TMDNA Preparation and Multiplex PCRKit、TransK RPlus DNAMaker均购自北京全式金生物技术有限公司,序列测定、全部引物均由南京金斯瑞生物科技有限公司合成,2×TaqMaster-Mix购自北京康为世纪生物科技有限公司,琼脂糖凝胶回收试剂盒和质粒小量提取试剂盒均购自美国Omega公司,荧光素酶分析试剂盒Dual-Lu-ciferase R ReporterAssay System购自美国Pro-mega,miR-151-3pmimics和U6均由上海吉玛制药技术有限公司设计合成,miRNA反转录引物和实时定量引物均由广州锐博生物科技有限公司设计合成,Opti-MEM、LipofectamineTM2000购自美国Invitrogen公司。
PCR仪、iQ5 Multicolorreal-timePCR仪均购自美国Bio-Rad公司,CO2细胞培养箱购自美国Thermo公司,OlympusIX71倒置相差显微镜购自日本奥林巴斯公司,多标记微孔板检测仪VictorX5购自美国PerkinElmer公司。
1.2试验方法
1.2.1miR-151-3p保守性分析和靶基因的检索在上下载小鼠、大鼠、猪和人等miR-151-3p的成熟序列,用Mega软件构建系统发育进化树,对其进行保守性分析。
利用Targetscan等在线预测软件对小鼠miR-151-3p靶位点进行预测,结合小鼠肌肉发育以及癌症发生过程中起调控作用的表达谱数据,确定候选靶基因。通过RNAhy brid深入分析小鼠miR-151-3p与靶基因结合的二级结构,筛选出与miR-151-3p种子序列结合自由能较小的靶基因。
1.2.2靶位点的扩增与荧光素酶报告载体的构建野生型靶位点的扩增:通过PCR分别扩增出Akt3、Twist1和Cacna1g3′UTR中与miR-151-3p结合的序列。反应体系:2×Taq Master Mix100μL,靶基因前引物和后引物各5μL(表1),DNA 5μg,ddH2O70μL。反应条件:95℃预变性5min;35个循环(95℃10s,60℃30s,72℃30s);72℃延伸10min。PCR产物经回收后取1μL与4μLpMD19-T载体按照说明书与1μLSolutionⅠ和5μL连接Buffer混成10μL体系。16℃,连接30min后,将连接产物转化涂板。37℃培养12h后,挑取单克隆摇菌,通过PCR和测序鉴定阳性克隆。
突变型靶位点的扩增:以上述阳性质粒为模板,分别用靶基因3′UTR的前引物与结合位点突变的后引物、结合区域突变的前引物与靶基因3′UTR的后引物组合扩增相应片段(表1),再将两段含突变序列的片段各取5μL混匀作为模板,用靶基因前后引物扩增。
PCR产物经胶回收后,连接至pMD19-T载体,转化涂板,测序鉴定种子序列有突变的克隆。荧光素酶报告载体构建:将与pMD19-T载体相连的含野生型或突变型靶基因3′UTR的质粒及报告载体psiCHECKTM-2分别经XhoⅠ、NotⅠ双酶切。双酶切体系:
20μL10×HBuff-er、20μLφ=0.1%的TritonX-100、20μLφ=0.1%的BSA、10μLXhoⅠ、10μLNotⅠ和10μg质粒,37℃恒温酶切16h,胶回收。分别取4μL目的片段和1μL报告质粒回收产物与1μLT4 DNA Ligase、2.5μL10×T4DNA Ligase Buffer和ddH2O混成25μL体系,16℃连接12h。最后,将连接产物转化涂板,PCR、测序鉴定阳性克隆。
1.2.3细胞培养及转染细胞培养:用φ=10%胎牛血清的DMEM高糖生长培养基在37℃、φ=5%CO2的条件培养C2C12。当细胞密度达到90%时,更换φ=2%马血清的DMEM分化培养基诱导分化。HEK293T细胞用φ=10%胎牛血清的DMEM生长培养基维持生长。
细胞转染:将C2C12以每孔2×106的数量接入12孔板中培养,在转染时融合度达70%。
按照lip of ectamine TM2000说明书,用Opti-MEM分别稀释脂质体、miR-151-3pmimics和阴性对照(NC),保持miR-151-3pmimics与NC终浓度均为100nmol/L,静置5min后,将稀释好的miR-151-3pmimics和NC分别与脂质体混匀,静置20min后,将转染混合物均匀滴加到细胞中,4~6h后换生长培养基。报告载体转染时,将HEK293T以每孔1.5×106的数量接种至24孔培养板。miR-151-3pmimics及NC的转染终浓度为50nmol/L,均以每孔100ng共转染野生型(psi CHECKTM-2-Target)或突变型(psi-CHECKTM-2-mTarget)质粒。
4~6h后,更换生长培养基。

1.2.4总RNA提取及实时定量PCR收集细胞:吸去培养基后用PBS洗去死细胞;每孔加入500μLTrizol,将细胞裂解物转移至1.5mL的离心管,室温静置5min;4℃、12000r/min离心10min;吸取上清液至新的1.5mL的离心管;加入100μL氯仿,剧烈振荡15s,静置5min;4℃、12000r/min离心15min;吸取上层水相转移至新的1.5mL离心管,加入等体积冰异丙醇,冰上沉淀10min;4℃、12000r/min离心10min,弃上清;用φ=70%冰乙醇洗涤沉淀,去乙醇;用DEPC水溶解RNA,测定浓度。


microRNA特异性反转录:取500ng总RNA,与2μL5×Primescript Buffer、0.5μL Primescript R Tase、0.8μLmi R-151-3p和U6特异性反转录引物混合进行特异性反转录。基因反转录依照说明书操作。反应条件均为37℃,15min;85℃,5s。实时定量PCR分析:microR-NA的反应以U6为内参,采用3步法:95℃,3min;45个循环(95℃,10s;60℃,20s;70℃,10s);溶解曲线设置为70℃,30s。基因以β-ac-tin为内参,反应条件为95℃,3min;45个循环(95℃,10s;60℃,30s);溶解曲线设置为55℃,30s。实时定量引物见表2。
1.2.5荧光素酶活性测定HEK293T细胞转染48h后,吸去培养基,用PBS清洗,加入100μL1×PassiveLysisBuffer,37℃恒温摇床上孵育15min充分裂解细胞;吸取20μL细胞裂解液至酶标板,加入100μL荧光素酶测试试剂Ⅱ(LARⅡ),混匀后测得内参萤火虫荧光素酶的活性。加入100μL Stop & Glo试剂,湮灭萤火虫荧光素酶反应,同时激活海肾荧光素酶反应,并立即检测海肾荧光素酶的活性。海肾荧光与萤火虫荧光比值即为相对荧光素酶活性。
1.3统计分析
每组试验重复3次,所有数据结果均用“平均数±标准误(mean±SEM)”表示。采用SPSS18.0软件进行独立样本t检验以比较处理组和对照组的差异(*表示P<0.05,**表示P<0.01)。
2、结果与分析
2.1进化树构建和靶基因预测
通过对多个物种系统发育分析发现,miR-151-3p成熟序列在大鼠和小鼠中高度保守,在人、猪等物种中同样具有高度保守性,与鼠类分布于NJ进化树的不同亚支(图1)。运用多个生物在线软件同时预测小鼠miR-151-3p靶基因,筛选出3个与肌肉发育和肿瘤发生相关的基因:Akt3、Twist1和Cacna1g。结果显示,Akt33′UTR的第208~215位、Twist13′UTR的第73~79位以及Cacna1g3′UTR的第463~469位均存在碱基序列CAGUCUA能够与miR-151-3p的种子序列GUCAGAU互补配对。
利用miRNA-mRNA互作软件RNAhy brid分析3个靶基因与miR-151-3p成熟序列结合的二级结构(图2),miR-151-3p与Akt33′UTR、Twist13′UTR和Cacna1g3′UTR结合的最小自由能(MFE)分别为-83.381、-101.817和-91.761kJ/mol。从理论上讲,miR-151-3p与Akt3、Twist1和Cacna1g的3′UTR均有稳定结合的可能。
2.2实时定量
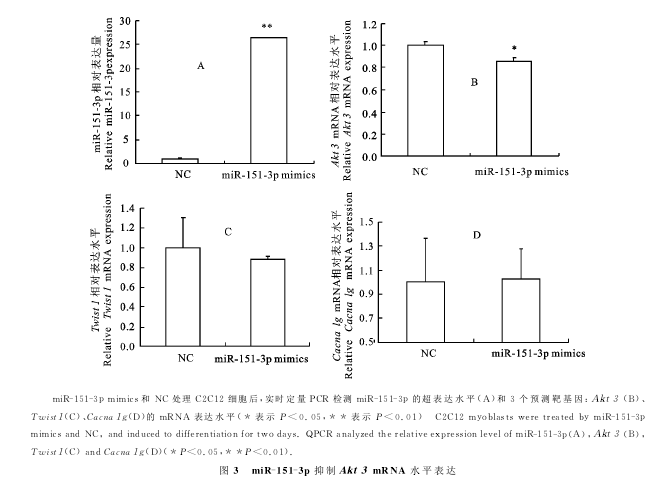
PCR检测靶基因mRNA表达水平将NC与miR-151-3 pmimics分别转染C2C12,分化2d后,收集细胞进行实时定量PCR检测。结果显示,与对照组相比,miR-151-3 pmimics处理组的miR-151-3p表达水平增高约26倍,证实miR-151-3 pmimics能显著提高miR-151-3p的表达(图3-A)。为进一步探究miR-151-3p超表达后相应预测靶基因的变化情况,对3个预测靶基因mRNA表达水平进行检测(图3-B、图3-C和图3-D),miR-151-3 pmimics处理组的Akt3mRNA水平与对照组相比降低约20%,表明超表达miR-151-3p能够显著抑制Akt3的mRNA水平;但超表达miR-151-3p后,Cacna1g和Twist1的mRNA水平与对照组相比并没有显著性的变化。初步证实Akt3是miR-151-3p的靶点之一。

2.3双荧光素酶报告载体的构建
双荧光素酶报告载体的构建模式见图4,将靶基因3′UTR插入psiCHECKTM-2中编码海肾荧光基因后的多克隆位点,构成靶基因的报告载体。提取C2C12细胞基因组DNA,用带XhoⅠ、NotⅠ限制性酶切位点的PCR引物,分别扩增含miR-151-3p结合位点的Akt3、Twist1和Cacna1g3′UTR的目的序列,扩增片段大小分别为186、228和260bp,序列见图5。胶回收目的片段后,利用分子克隆技术将扩增片段连接至pMD19-T载体。以鉴定正确的质粒为模板,采用重叠PCR技术,分别进行扩增。
3个靶基因各获得两段带有突变位点的扩增片段(图6-A和图6-B)。将分段PCR产物用野生型引物进行重叠PCR扩增,获得的PCR产物经凝胶电泳,在目的大小处可见1条清晰条带(图6-C)。回收目的片段连接至pMD19-T载体,获得3个基因3′UTR与种子序列结合位点突变的载体。
选择测序正确的靶基因3′UTR野生型和突变型TA质粒与psi CHECKTM-2质粒,分别经XhoⅠ、NotⅠ双酶切后,连接,转化至感受态,PCR和测序结果显示野生型和突变型双荧光素酶报告载体构建成功。将靶基因野生型报告载体分别命名为psiCHECKTM-2-Akt3、psi-CHECKTM-2-Twist1和psiCHECKTM-2-Cacna1g,突变型报告载体分别命名为psiCHECKTM-2-mAkt3、psiCHECKTM-2-mTwist1和psi-CHECKTM-2-mCacna1g。
2.4双荧光素酶报告系统验证
miR-151-3p靶基因提取miR-151-3p靶基因野生型和突变型报告质粒,将psiCHECKTM-2-Akt3、psiCHECK-TM-2-mAkt3、psiCHECKTM-2-Twist1、psi-CHECKTM-2-mTwist1、psiCHECKTM-2-Cac-na1g和psiCHECKTM-2-mCacna1g质粒分别与miR-151-3pmimics或NC共转染至HEK293T细胞,培养48h后裂解细胞,用双荧光素酶分析试剂盒检测海肾和萤火虫荧光信号的强度(图7)。当共转染靶基因野生型报告载体与miR-151-3pmimics时,miR-151-3p能够显著抑制含miR-151-3p结合位点的Akt3、Twist1和Cacna1g报告载体的荧光素酶活性,3个预测靶基因的相对荧光素酶活性与对照组相比分别下降50%、40%和30%。然而,共转突变型报告载体、miR-151-3pmimics或NC后,除Akt3原本受到抑制的相对荧光素酶活性可以通过3′UTR中miR-151-3p结合位点的突变得到恢复外(图7-A),其他两组中,Cacna1g和Twist1的突变型报告载体的相对荧光素酶活性相较于对照组仍然保持着显著抑制的状态,与转染了野生型报告载体的结果相比没有差异(图7-B和图7-C)。再一次验证Akt3是miR-151-3p的靶位点。

3、讨论
miRNAs是一类进化保守的内源性小分子RNA。miRNAs基因通过RNA聚合酶Ⅱ转录出含有5′帽子结构和3′多聚腺苷酸尾巴的pri-miR-NAs。
pri-miRNAs经过一系列切割、运输、释放,最终形成长度约为22nt的双链,成熟的miR-NAs分子被解链,单链miRNAs进入核糖核蛋白复合体。在哺乳动物中,miRNAs主要通过调控靶基因表达从而影响基因作用。目前有关miRNAs的报道涵盖了包括增殖、分化、再生等生物学领域。

据文献报道,miR-151-5p具有癌基因的作用,但共转录的miR-151-3p仅在肾病综合征中有报道表达增高,对肿瘤发生和肌肉发育等功能的研究还未见报道。本研究以miR-151-3p为研究对象,采用软件对miR-151-3p成熟序列进行保守性分析发现,miR-151-3p成熟序列在脊椎动物中进化保守性较高,小鼠和人的成熟序列仅在非种子序列存在一个碱基的差异,两者在种子序列与靶基因的结合上可能具有相似性。当前大多数有关miR-151的研究集中在人类肿瘤细胞,因此,推断小鼠miR-151-3p也可能在肿瘤发生中起作用。
为了进一步阐明miR-151-3p作用机制,通过TargetScan和PicTar等搜索并预测了小鼠miR-151-3p的靶基因。在重叠的候选基因中,能通过PI3K依赖性的机制调节细胞生存和癌症发生的Akt3,影响肿瘤细胞凋亡的癌基因蛋白Twist1和钙离子通道相关蛋白Cacna1g被认为是miR-151-3p可能的靶基因,并且3个靶基因与miR-151-3p结合区在脊椎动物中的保守性较高。采用RNAhybrid软件对miR-151-3p与3个靶基因3′UTR局部互补的二级结构进行预测,最小自由能越小,则结合越紧密,结构越稳定。3个靶基因都参与肿瘤细胞的病变等过程,提示miR-151-3p可能通过这3个靶基因调控肿瘤和肌肉发育过程。

为证明miR-151-3p能够靶向调节Akt3、Twist1和Cacna1g,用miR-151-3pmimics转染C2C12后检测3个预测靶基因的变化情况,过表达miR-151-3p能显著抑制Akt3的mRNA水平,而对Twist1和Cacna1g的表达并没有影响,这些结果暗示Akt3而不是Twist1和Cacna1g受miR-151-3p的调节。为进一步验证该结果,通过构建预测靶基因的双荧光素酶报告载体,将3个靶基因含miR-151-3p结合位点的3′UTR序列及结合区域突变的片段连接到报告载体psi-CHECKTM-2,进一步利用双荧光素酶报告基因检测系统研究miR-151-3p与预测靶位点3′UTR是否存在互作,结果显示,在Akt3与miR-151-3p种子区域结合的序列突变后导致miR-151-3p对海肾荧光素酶活性的抑制由野生型的50%降至20%,而共转Twist1或Cacna1g的野生型或突变型报告载体与对照组相比都显示出miR-151-3p对海肾荧光素酶活性的显著抑制。由于miRNAs与靶基因的结合并不需要种子序列的完全匹配,而现有的生物软件并不能预测这种结合位点。
Twist1和Cacna1g在软件预测结合位点处可能并没有与miR-151-3p结合,而在扩增的3′UTR片段中还存在其他位点与miR-151-3p结合,还需要进一步试验验证。综上所述,Akt3是miR-151-3p的靶基因,能被miR-151-3p直接调控,为深入研究miR-151-3p的功能提供新的理论依据。





