摘 要: 为研究复制性衰老中表观遗传学的变化,以年轻代龄(PD26)和年老代龄(PD55)的人胚肺二倍体成纤维细胞(2BS)为材料,进行ATAC-seq测序,并使用Bowtie2、FastQC、MACS、deepTools、phastCons、R语言、HOMER以及基因本体论(Gene Ontology, GO)对测序结果进行深入分析和挖掘。结果显示, 2BS细胞染色质开放区域的保守性较强,其主要集中于启动子(promoter)区至转录起始位点(transcription start site, TSS),且年轻细胞与年老细胞的启动子区至转录起始位点的开放程度不同,细胞随着衰老在启动子区至转录起始位点的开放程度逐渐下降。进一步对年轻和年老细胞之间存在显着差异的靶基因进行GO分析发现,这些差异基因主要与细胞进程、代谢、生物性调节、结合功能、催化功能相关。本次研究发现衰老过程中染色质开放区减少、转录调控水平下降,提示复制性衰老与转录调控关系密切。
关键词: 复制性衰老; ATAC-seq测序; 转录调控; 染色质开放程度;
Abstract: To study the epigenetic changes in replicative senescence, the ATAC-seq was performed in young(PD26) and senescent(PD55) 2BS cells. After sequencing, the results were analyzed by Bowtie2, FastQC,MACS, deepTools, phastCons, R language, HOMER and Gene Ontology(GO). The enrichment sites of open chromatin regions were highly conserved. Although the open regions were mainly enriched between the pro-moter and transcription start site(TSS), the enrichments in the young and the old are not the same, varying with aging of 2BS cells. Furthermore, GO analysis was performed to analyze the differentially expressed genes in different ages of 2BS cells. The genes were mainly clustered in cellular process, metabolic process,biological regulation, binding and so on. These results showed that the aging process was accompanied by declined degree of chromatin openness and decreased level of transcriptional regulation, which revealed the close relationship between the replicative senescence and transcriptional regulation.
Keyword: replicative senescence; ATAC-seq; transcriptional regulation; opening of chromatin;
复制性衰老又称细胞衰老,当细胞周期停滞、凋亡抵抗以及部分基因如P16INK4a表达改变时,细胞出现衰老表型[1,2]。复制性衰老是整体衰老的基础,近期研究发现其具有拮抗癌症、使癌细胞得以清除的能力[3,4]。近年来,随着人口老龄化的发展及癌症等老年病发病率的增加,细胞衰老作为老年疾病的主要病理基础,已成为分子生物学研究的热点之一[5,6,7,8]。
在真核生物中,DNA围绕组蛋白分级折叠,形成核小体,而核小体则通过进一步紧密折叠形成高级结构———染色质[9]。此时,根据细胞所处的状态,其基因组可划分为活跃表达的基因与相对沉默的基因。在DNA分级折叠的这一过程中,非活跃基因表达相关的结构区域相对封闭、核小体结构致密,呈转录抑制状态;而与活跃表达基因相关的区域则呈开放状态,核小体相对松散,有利于转录因子、组蛋白等调节元件与这些基因的启动子、增强子相互作用,促进转录的进行[9,10]。通常情况下,染色质的开放程度可代表转录因子、组蛋白等调节元件与DNA的结合状态,因此根据染色质的开放程度明确生理学状态、疾病发生发展中表观遗传学状态的改变,也已成为表观遗传学技术的主要切入点[9,11,12,13]。

迄今为止,细胞衰老过程中表观遗传学状态的改变尚不明确,从染色质空间结构、基因组学角度对染色质开放性的变化进行分析探讨的研究较少,在细胞衰老过程中染色质开放程度的具体变化仍未知[14,15]。故本研究采用基于转座酶Tn5和高通量测序的染色质开放性分析技术ATAC-seq(assay for transposase-accessible chromatin with highthroughput sequencing),研究在复制性衰老过程中全基因组染色质开放程度的变化,为探索复制性衰老过程中表观遗传以及转录调控水平的变化提供初步的研究基础。
1、 材料和方法
1.1、 材料
年轻代龄(PD26)和年老代龄(PD55)的人胚肺二倍体成纤维细胞(2BS)购自武汉大学中国典型培养物保藏中心。
1640培养基、胎牛血清(fetal bovine serum,FBS)、青霉素链霉素混合液和磷酸盐缓冲液(phosphate buffer saline,PBS)购自美国Gibco公司;Tris、NaCl以及MgCl2购自BBI生命科学有限公司;Tn5转座酶源自美国Illumina公司;NP-40和nuclease-free H2O购自上海碧云天生物技术有限公司;DNA纯化试剂盒源自美国Qiagen公司;PCR扩增试剂盒源自美国New England BioLabs公司。
二代测序仪(150PE,Illumina公司,美国);qPCR仪(480,Roche公司,瑞士);低温高速离心机(CR3I,Thermo公司,美国)。
1.2 、Tn5酶切反应
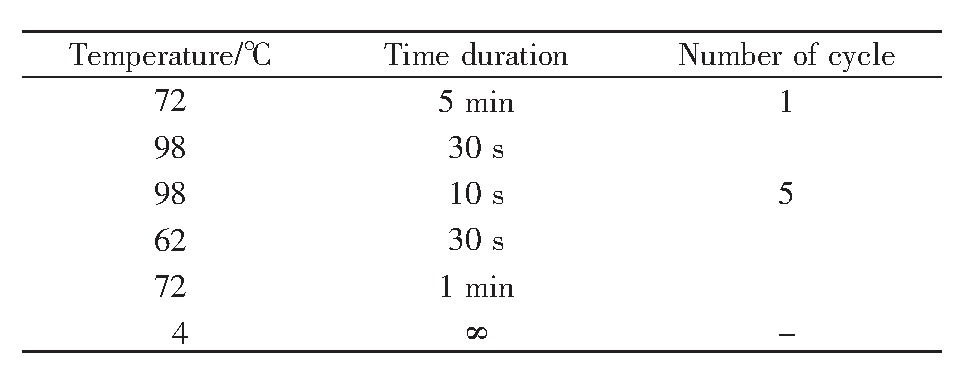
参照文献[16]的方法,对2BS细胞进行Tn5酶切反应,主要步骤如下:取冻存于液氮中的PD26、PD55的2BS细胞进行复苏。复苏后的细胞置于T75培养瓶,采用添加10%胎牛血清、1%青霉素链霉素混合液的1640培养基培养,培养箱条件为37℃、5%CO2。当细胞长满且生长状态良好时,用胰酶将细胞消化,并使用台盼蓝染色,过滤除去死细胞后进行细胞计数,计数50 000个细胞。取计数后的细胞,在4℃条件下500 g离心5 min后弃上清;再加入50μL预冷PBS吹打洗涤,4℃条件下500 g离心5 min,弃上清;加入50μL预冷的裂解液(由10 mmol/L Tris pH 7.5、10 mmol/L NaCl、3 mmol/L MgCl2以及0.1%NP-40配置而成)悬浮细胞,在4℃条件下500 g离心10 min后弃上清。此时向细胞沉淀中加入50μL转座反应体系(表1),吹打混匀后,37℃孵育30 min。待孵育结束后使用DNA纯化试剂盒纯化DNA,纯化完成后使用10μL试剂盒中的洗脱液将其洗脱,随后进行PCR扩增。PCR反应体系如表2所示,循环条件见表3。其中,PD26对应的primer 1序列为AGGCAGAA,PD55对应的primer 1序列为TCCTGAGC;barcoded primer 2序列为AATGAT-ACGGCGACCACCGAGATCTACACTCGTCGGCA-GCGTCAGATGTG。PCR结束后,再次使用DNA纯化试剂盒纯化DNA[17]。
表1 转座反应体系
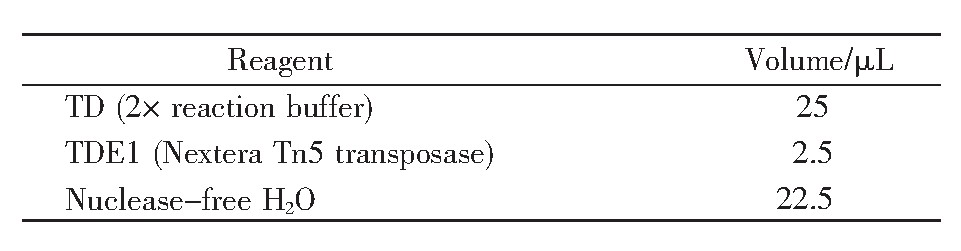
表2 PCR反应体系

1.3、 ATAC-seq测序、质量评估及原始数据过滤
使用Illumina Hiseq 150PE测序仪对Tn5酶切反应得到的DNA进行双端测序,识别chasity>0.6的碱基,对每个测序序列(sequenced read)进行pass filter检验(当某一read的前25个碱基中,仅存在一个或者不存在chasity≤0.6的碱基时,判定该read符合检验标准),过检后的单克隆reads则使用FastQC进行测序质量评估。判定评估合格的标准如下:每个碱基的质量均≥5、每组碱基的中位数均≥20;tile的质量检测中不存在暖色结果;序列质量评分均集中于高分;GC碱基含量分布均匀;GC碱基占比均匀;测序中序列长度分布较一致;测序序列中接头(adapter)出现频率较低;测序序列中特征性短序列重复出现的次数较低。如果评估合格,则进行数据过滤,去除低质量(QPhred≤20的碱基数占整个read长度50%以上的reads)、N(N表示无法确定碱基信息)的比例>10%和包含adapter的reads,以保证分析质量。
表3 PCR循环条件
1.4、 ATAC-seq的序列比对和结合位点(peak)扫描
使用Bowtie2(版本2.25)对参考基因组(hg19)建立索引回帖后,进行序列比对。然后根据泊松分布,使用MACS(版本2.1.0)对测序后各个区域基于唯一比对的read数的P-value值进行计算,当P-value<1.0E-05时,该区域则被认为是一个结合位点(peak)。标准化后的P-value值越高,理论上该区域的染色质越致密[18]。
1.5、 保守性分析
使用phastCons软件,根据phylo-HMM模型对结合位点的保守性进行计算[19]。
1.6 、回帖序列在基因附近的信号分布情况分析
将所有基因作为目标区域,使用deepTools对基因长度(转录起始位点TSS到转录终止位点TTS之间的距离)进行归一化处理,绘制ATAC-seq reads(使用bigwig文件)在转录起始位点上游3 kb到转录终止位点下游3 kb范围内的平均信号值,以average plot的形式将结果展示。
1.7、 结合位点注释分析
使用HOMER软件的annotatePeaks.pl工具寻找离每个结合位点距离最近的转录起始位点所属的基因(以下称为结合位点邻近基因),以及结合位点覆盖的基因,并注释其覆盖的功能性区域。注释的功能性区域包括启动子到转录起始位点的区域(promoter-TSS,默认范围为基因起点上游1 kb到基因起点下游100 bp)、转录终止位点(TTS,默认范围为基因终点上游100 bp到基因终点下游1 kb)、外显子(exon)、内含子(intron)和基因间区(intergenic)。如果一些功能性区域有重叠部分,则按上述顺序优先选择排在前面的功能性区域作为最终注释。对上述注释分析结果中结合位点在所覆盖基因的功能性区域上的分布进行统计,并使用R语言的ggplot2包将统计结果绘制成饼状图和柱状图。
1.8、 差异结合位点分析
使用bedtools工具将2BS-PD55与2BS-PD26的peak文件合并,并对合并文件进行处理。如果两个结合位点有重叠区域,则合并成一个新的结合位点。计算PD26与PD55在每个合并的新结合位点区域上的read数目,然后使用DEGseq进行差异分析,得到样本间的差异结合位点,即差异染色质开放区域。本次差异结合位点的筛选原则为|log2FC|>1且P-value<0.05。
1.9、 差异结合位点注释分析
使用HOMER软件的annotatePeaks.pl工具寻找离PD26与PD55差异结合位点距离最近的转录起始位点所属的基因,以及差异结合位点覆盖的基因,并注释其覆盖的功能性区域。对差异结合位点注释分析结果中结合位点在所覆盖基因的功能性区域上的分布进行统计,并使用R语言ggplot2包将统计结果绘制成饼状图。
1.1 0 、GO富集分析
基因本体论(Gene Ontology,GO)数据库可对基因功能进行富集分析。我们使用GO Term Finder,将PD26与PD55差异结合位点临近的基因与GO term数据库比对,计算每个term的基因数量,然后应用超几何检验,找出整个基因组背景下结合位点相关基因中显着性富集的GO term,从而识别出差异结合位点相关基因行使的主要生物学功能。
2 、结果
2.1 、测序质量
2.1.1、 测序的错误分布率
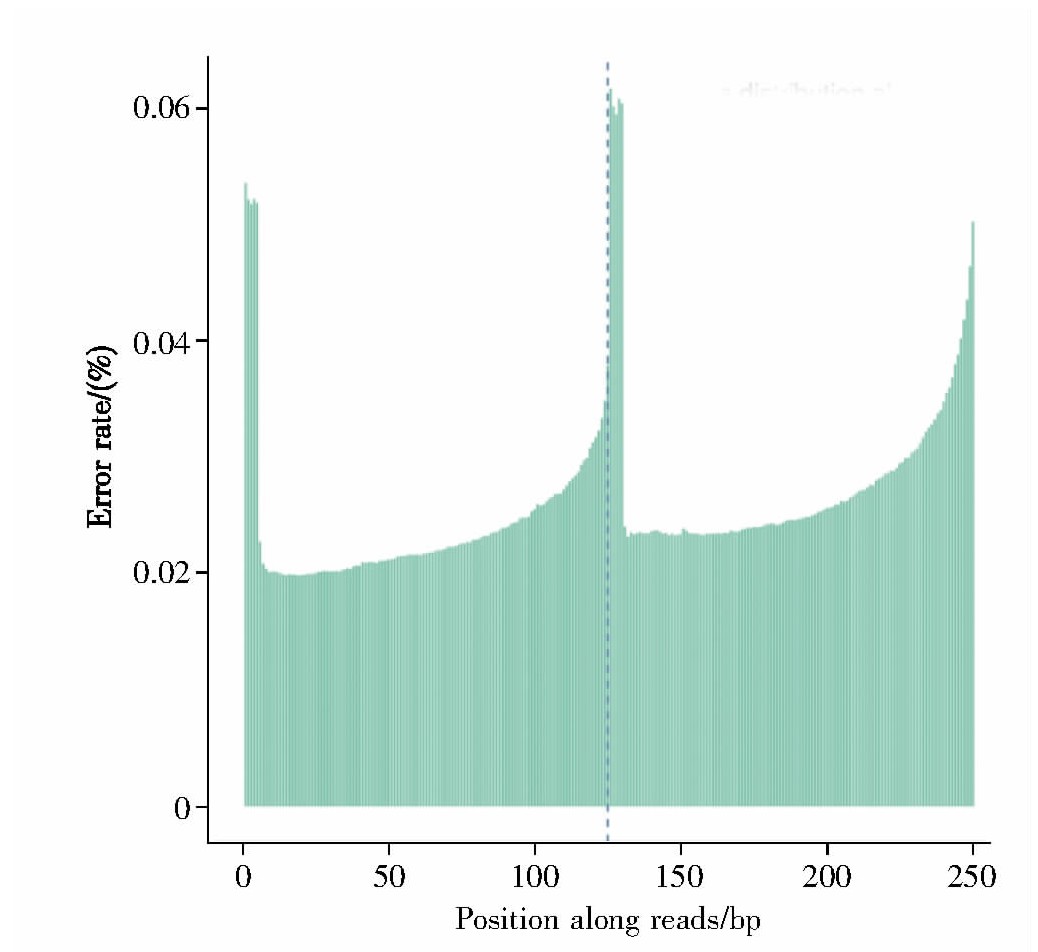
对Tn5酶切后的年轻与年老细胞中的DNA进行双端测序,排除酶切后碱基质量因素以及测序仪和测序试剂等的影响后,所得reads的测序错误率如图1所示,均低于0.06%。本实验测序的错误分布率检查合格、测序质量较高,提示此次测序结果可用于后续分析。
Fig.1 Error rate distribution along r
2.1.2 、碱基含量分布
使用FastQC对Tn5酶切后的年轻与年老细胞中的DNA进行碱基组成检测,结果如图2所示。各嘌呤碱基和嘧啶碱基的组成比例都较均衡,低质量碱基(指在某一read的某一位置中,含量小于20%的碱基)比例较低且仅出现于随机引物扩增的开始阶段,表明本次测序结果较好,可信度较高。
2.2、 酶切片段长度的分布
酶切片段(insert size,也称作fragment length)是指经转座酶Tn5切割后获得的DNA片段,常用于监测Tn5酶切反应中酶的用量是否合适。本实验的酶切片段长度分布如图3所示,从左至右依次为无核小体片段(nucleosome free,即开放染色质区域)、单核小体片段(包括一个核小体的酶切片段,长度一般在147 bp到147*2 bp之间)和双核小体片段(包括两个核小体的酶切片段)的长度分布,表明本次实验酶切效果较好。
2.3、 结合位点统计
对测序产生的reads的P-value值进行计算,通常标准化后的P-value值越小,理论上该区域的染色质越疏松[18],越可能成为染色质的开放区域从而结合转录因子。对本次测序样本的peaks进行统计分析后发现,PD26的2BS细胞有45 681个peaks,而PD55的2BS细胞有50 075个peaks。
2.4、 结合位点保守性分析
图4展示了年轻细胞与年老细胞进化保守性的分析结果。在脊椎动物基因组学中,调节复制、转录和翻译的序列,常被认为是功能性保守序列[20]。在本次实验中,我们对结合位点进行统计分析发现,年轻细胞与年老细胞的染色质开放性变化区域较保守、相似性高,因此可以初步推断这些结合位点的相关区域在转录表达中具有较高的重要性。
图2 测序序列碱基分布示意图
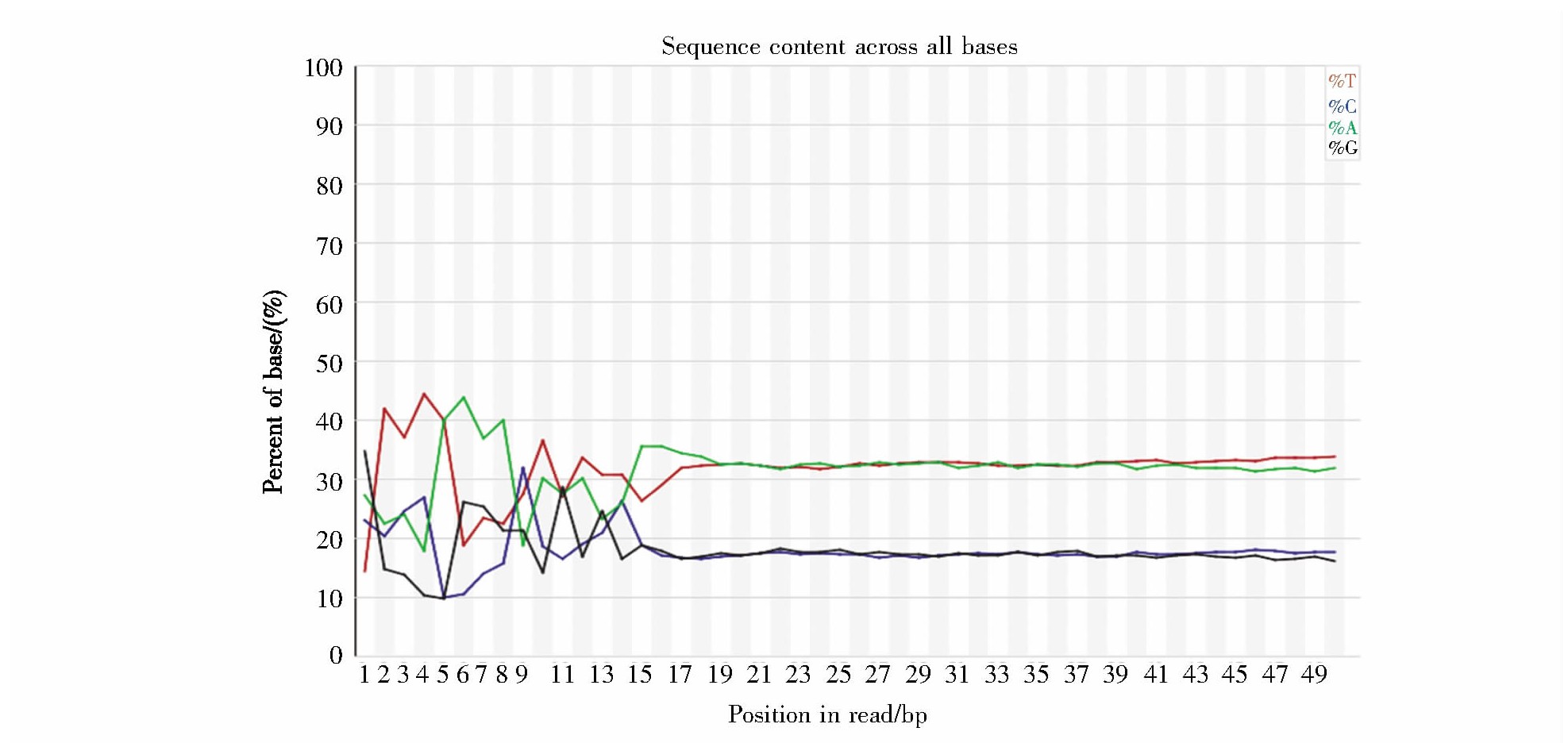
Fig.2 Sequence content across all bases
图3 酶切片段长度的分布
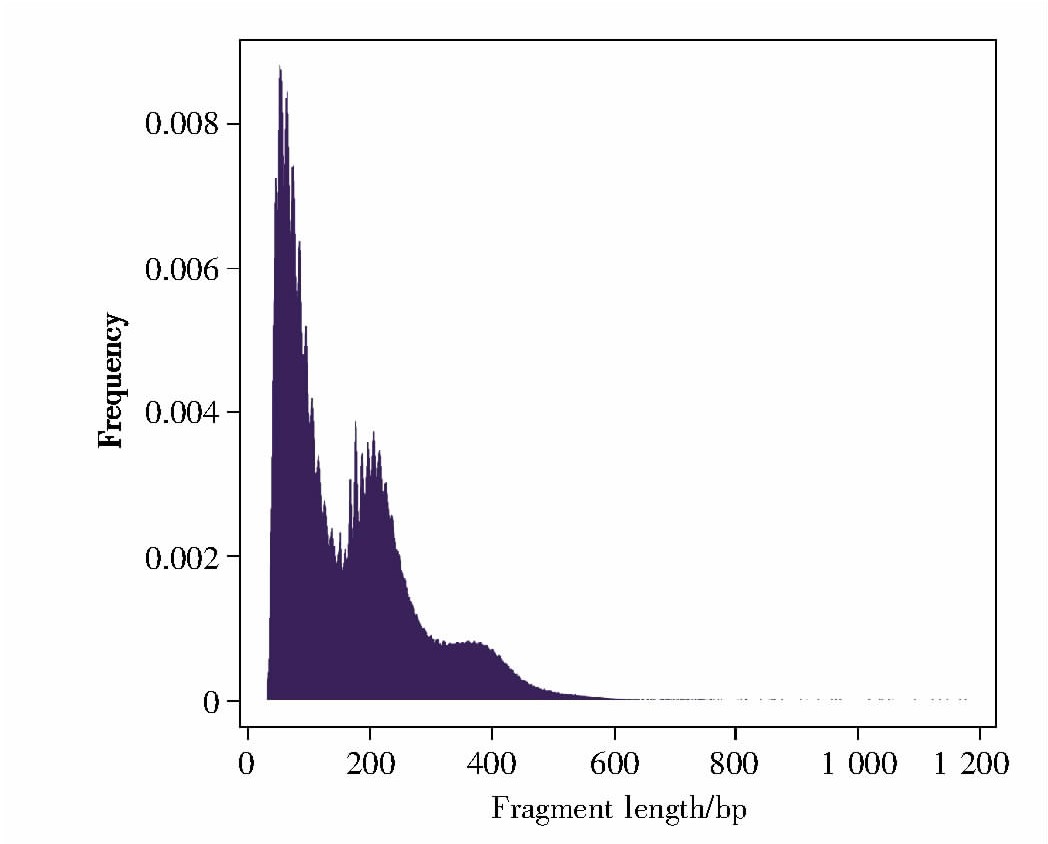
Fig.3 Fragment length distribution
X轴代表Tn5酶切后DNA片段的长度,Y轴代表该长度片段出现的比例。
The X-axis represents fragment length,and the Y-axis represents the frequency of length distribution.
图4 结合位点保守性分析
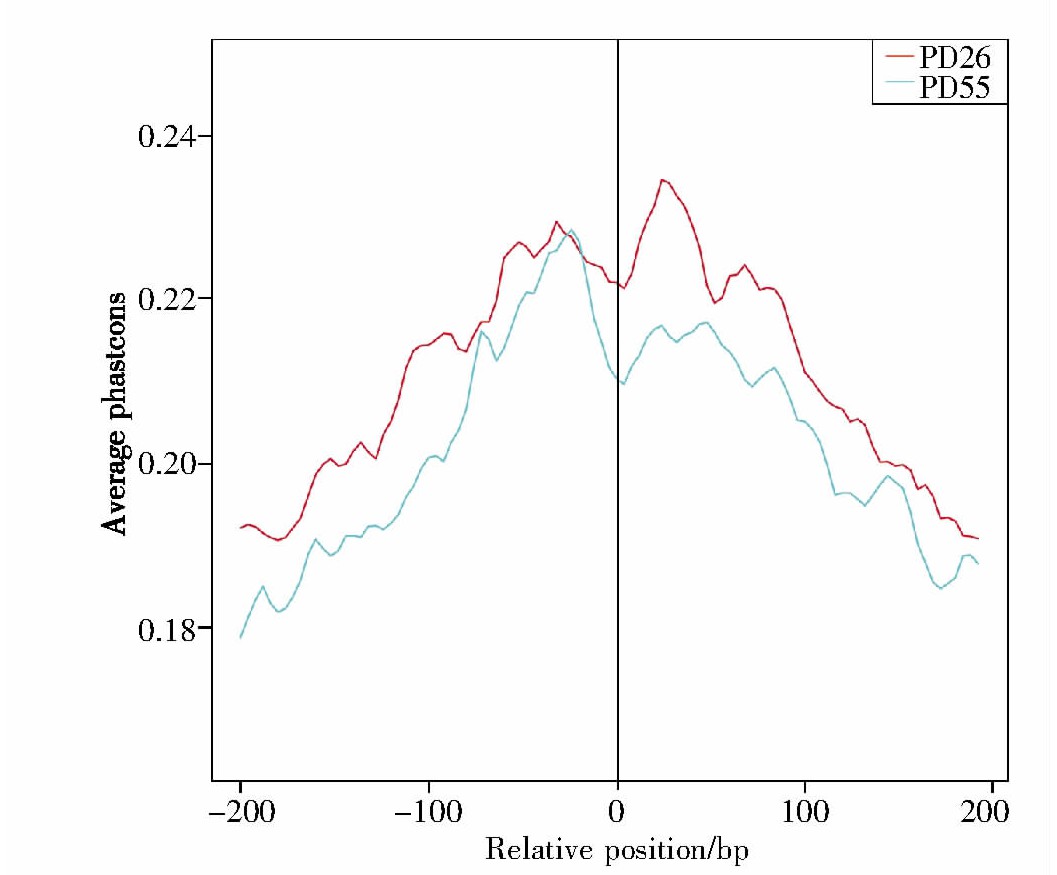
Fig.4 The results of conservation
X轴代表此次检测所得的结合位点对应基因组中该位点的相对位置信息,其中“0”表示相对位置的中点;Y轴代表保守性的评分,分值越高代表保守性越强。
The X-axis represents position information,and“0”represents relative position of peaks.The Y-axis represents conservation score(a higher score indicates a higher conservation).
2.5、 回帖序列(mapped reads)在基因附近的平均信号分析
在所有基因长度归一化后,将reads与hg19基因组比对,计算其在基因组上的分布情况,结果如图5所示。从结果可知,回帖序列(即染色质的开放区域)主要集中在转录起始位点附近,提示这些染色质的开放区域与活跃的转录调控相关。同时,我们发现,随着细胞衰老程度的加深,转录起始位点附近的染色质开放程度明显下降,提示随着细胞衰老的发生发展,转录水平进一步下降。
图5 回帖序列在基因附近的平均信号分布
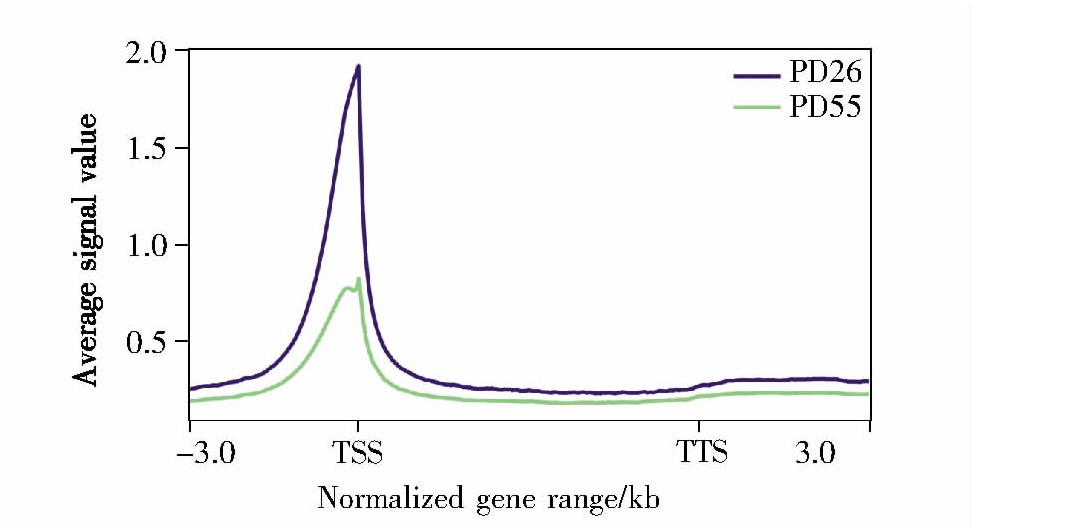
Fig.5 The relationship between mapped reads and genes in average plot
X轴表示归一化后的基因范围,“-3.0”表示TSS上游3 kb,“3.0”表示TTS下游3 kb;Y轴表示位点的平均信号,数值越大代表越富集。
The X-axis represents the normalized gene range,“-3.0”represents 3 kb upstream the TSS,and“3.0”represents 3 kb downstream the TTS.The Y-axis represents the average signal value of the site.The enrichment degree is proportional to the Y-axis value.
2.6、 结合位点在全基因组功能区上的注释
全基因组的功能区域分为启动子到转录起始位点的区域、转录终止位点、外显子区、内含子区和基因间区。转录因子一般集中分布在转录起始位置附近,即TSS区域。结合位点在全基因组功能性区域上的分布如图6所示,年轻细胞(2BS-PD26)、年老细胞(2BS-PD26)的染色质开放区段主要位于:内含子区、基因间区、启动子到转录起始位点的区域、外显子区和终止子。除去在全基因组中占比极大且不参与基因表达的内含子和基因间区后,染色质开放区主要富集于启动子到转录起始位点的区域。此外,图7结果显示年轻细胞与年老细胞中结合位点在该区域分布的比例分别为23.04%和15.4%。以上信息表明转录调控对复制性衰老有着重要影响,且随着细胞的逐渐衰老,启动子到转录起始位点区域的开放比例进一步下降,即转录的水平进一步下降。
2.7 差异结合位点统计
通过分析PD55与PD26之间的差异结合位点,可以得到衰老状态下特异性染色质开放区域。本次实验样本间的差异结合位点数目为15 812。
图6 结合位点在基因组功能性区域的分布统计图
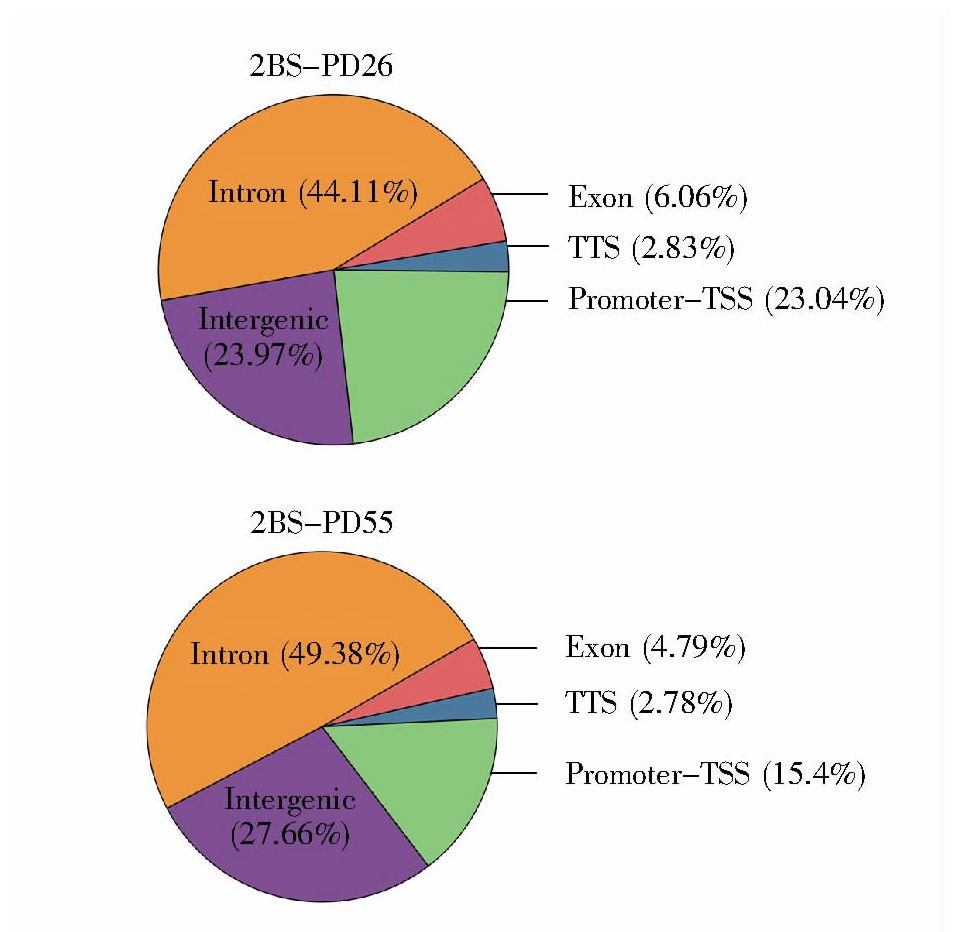
Fig.6 The annotation of peaks in the genome
图7 结合位点在启动子到转录起始位点区域的分布统计图
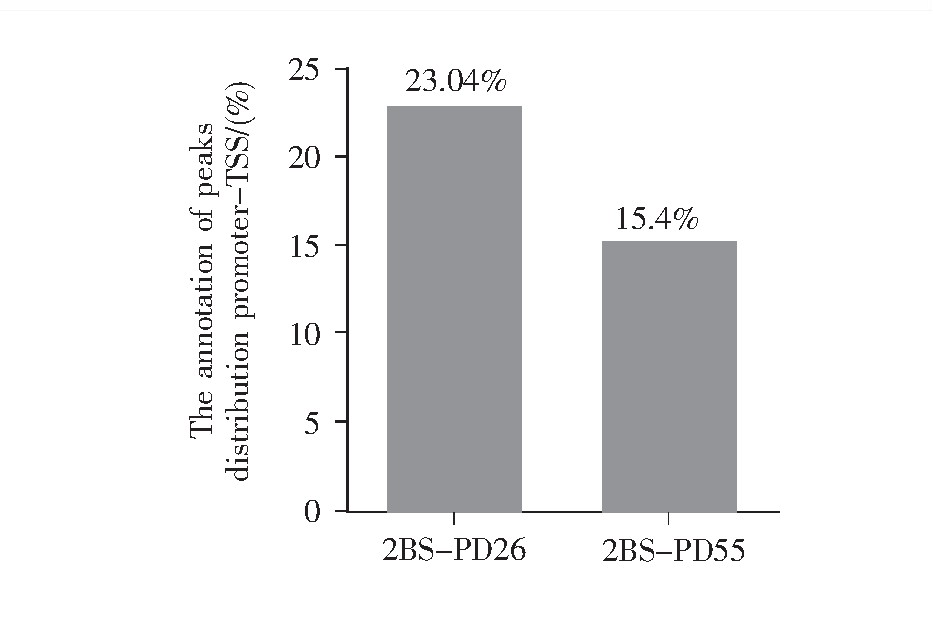
Fig.7 The annotation of peaks in promoter-TSS
2.8、 差异结合位点在全基因组功能区上的注释
2BS-PD26和2BS-PD55的差异结合位点在全基因组功能区的注释如图8所示。结果表明,细胞衰老后染色质关闭与开放的区域主要集中在内含子区和启动子到转录起始位点区域。其中,染色质关闭的区域有40.31%分布于内含子区,有26.45%分布于启动子到转录起始位点区域;染色质开放的区域有52.55%分布于内含子区,有6.18%分布于启动子到转录起始位点区域。
同时,在注释中我们可以发现基因间区占比较多,推测可能因为基因间区在基因组上的占比较大,故检测到的结合位点相对其他区域更多,但此类结合位点并非真正的转录调控因子的结合位点。
图8 差异结合位点在基因组功能性区域的分布统计图
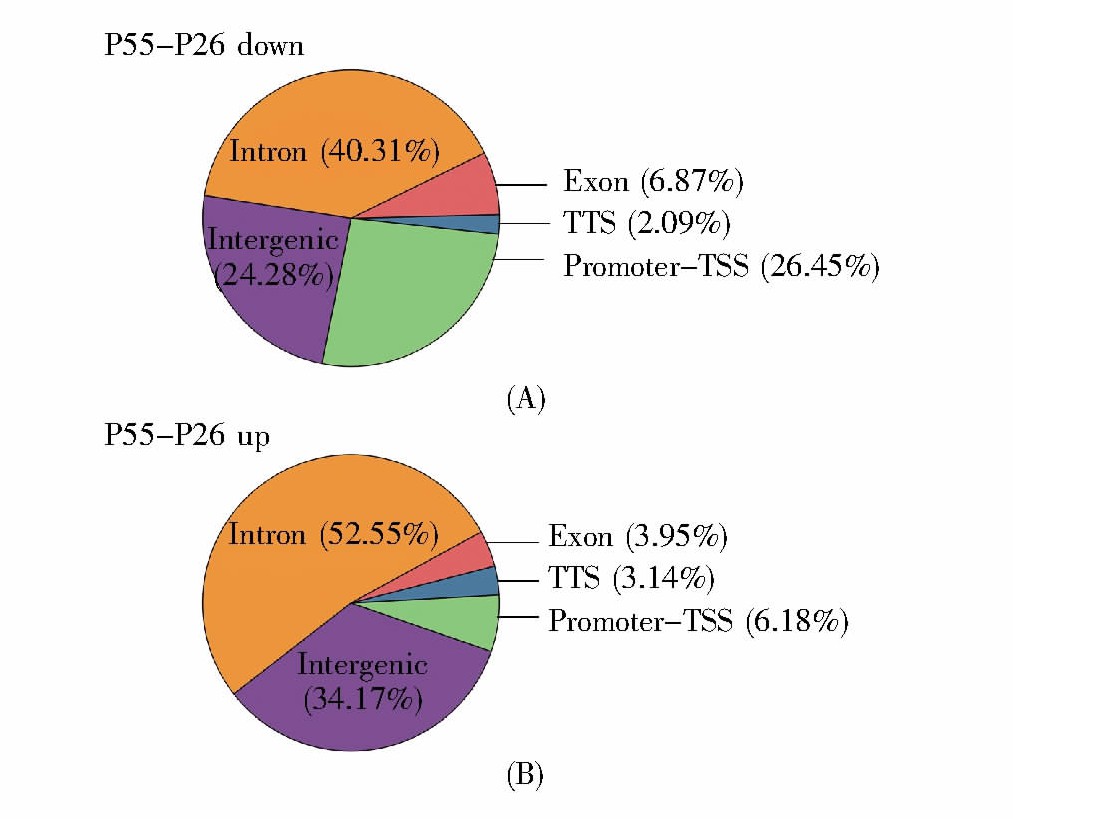
Fig.8 The annotation of different peaks in the genome
(A)细胞衰老后,染色质关闭区域在基因组功能性区域的分布;(B)细胞衰老后,染色质开放区域在基因组功能性区域的分布。
(A)The distribution of closing chromatin regions’annotation;(B)The distribution of open chromatin regions’annotation.
2.9、 差异位点靶基因的GO分析
对2BS-PD26和2BS-PD55的每个差异结合位点的邻近基因进行GO注释,部分结果如表4所示。
表4 差异位点靶基因GO富集分析

图9 差异位点靶基因的GO富集分析
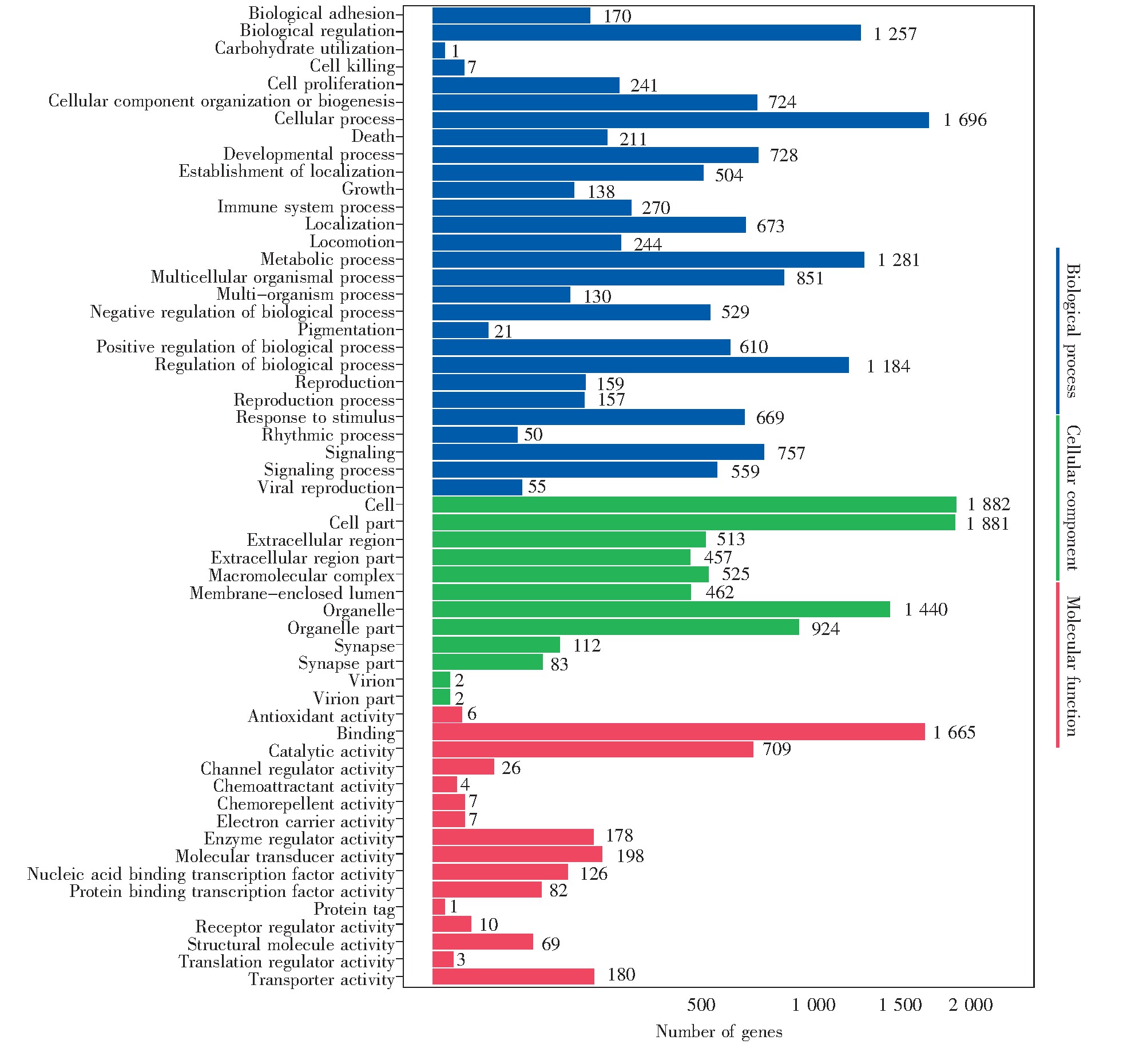
Fig.9 GO term enrichment analysis to the target gene of different peaks
选取差异位点邻近基因显着富集的GO条目绘制柱状图,结果如图9所示。富集程度较高的有细胞进程(cellular process)、代谢(metabolic process)、生物性调节(biological regulation)、结合功能(binding)、催化活性(catalytic activity)等,表明复制性衰老与细胞自身调节紧密相关,而这些生物学过程,均与转录调控有密切联系。
3、 讨论
尽管目前针对复制性衰老过程中表观遗传学变化的研究方法有很多,如使用DNA酶1酶切基因组后进行高通量测序的DNase-seq、使用微球菌核酸酶酶切基因组后进行高通量测序的MNase-seq和利用甲醛对染色质固定后进行高通量测序的FAIRE-seq,但此类方法对实验组细胞数量要求极高、所需测序深度较高、实验步骤繁琐且可重复性极低[10,16]。ATAC-seq的开发与应用,极大程度减少了样本的使用量、增加了实验的可重复性[21]。本实验中的测序质量分析显示:碱基错配率极低、碱基含量分布均匀,这再次证明该实验方法的准确性。同时,ATAC-seq作为检测表观遗传学变化的一种新方式,可对靶基因的表观状态给出更为准确的推测,具有一定的可挖掘性[22,23]。
本研究通过ATAC-seq技术结合生物信息学分析方法,对年轻代龄(PD26)和年老代龄(PD55)的2BS细胞展开了分析,初步发现,2BS细胞中结合位点的基因保守性均较强;ATAC-seq的回帖序列主要富集于转录起始位点附近;结合位点特异性地注释于启动子到转录起始位点区域,提示复制性衰老与转录调控具有较强的相关性。
文中在分析回帖序列在基因附近的平均信号分布时,发现染色质的开放区域主要富集在转录起始位点附近,且年轻细胞与衰老细胞在转录起始位点的开放程度不同,衰老时染色质的开放程度明显下降。这可能与衰老时基因的表达水平改变相关,衰老时细胞内各类蛋白质的表达大部分下降。本实验室前期研究发现,E2F1、POLD1等基因的表达水平随着细胞衰老出现下降的趋势[24,25,26],此类基因的启动子区在年轻细胞中呈开放状态,当细胞衰老时则表现得较为封闭。故此类基因的回帖序列经过富集分析,可出现衰老时转录起始位点的富集减少。但是,转录起始位点区域富集减少并非意味着衰老时细胞不出现转录调控。此时,细胞内以E2F1、POLD1为代表的一类基因还可以出现微弱表达,而另外一些基因(如P16INK4a等)则随着细胞衰老的出现表达上升[27,28],这些表达上调基因的启动子区会随衰老的发生发展进一步开放,从而使转录起始位点富集的回帖序列增多。由于衰老时呈表达增多的基因仍较表达减少的基因少,所以从全基因组的层面分析,衰老时染色质的开放区域在转录起始位点区的富集下降。
其实,结合位点在全基因组功能区上的注释,也进一步阐明了这一问题。对于年轻与年老的2BS细胞,其开放区段均富集于启动子到转录起始位点区域,表明转录调控对复制性衰老有着重要影响。随着细胞的逐渐衰老,启动子区的开放比例进一步下降,说明当细胞衰老发生时,细胞内的转录调控水平减弱,而在年轻细胞中转录调控较为活跃。当然,在年老细胞中也存在开放的启动子区,这是因为衰老的细胞也存在需要转录表达的基因。
此外,我们使用GO富集分析进一步验证了转录调控对复制性衰老的调节。其中,涉及细胞进程、代谢、生物性调节、结合功能、催化活性等功能的基因,在细胞衰老过程中富集,此类基因与细胞自身调节、转录调控均有密切联系。同时,我们也发现,作为复制性衰老经典标志物的P53、P16INK4a[1,29,30]等,均在GO分析结果中富集,如P53富集于GO:0048522、GO:0071840、GO:0016043等;P16INK4a富集于GO:0071840、GO:0016043、GO:0044260等。
综上所述,从转录调控探究、理解复制性衰老,应为我们后续研究的主要工作之一。
参考文献
[1]LOPEZ-OTIN C,B LASCO M A,PARTRIDGE L,et al.The hallmarks of aging[J].Cell,2013,153(6):1194-1217.
[2]CHILDS B G,BAKER D J,KIRKLAND J L,et al.Senescence and apoptosis:dueling or complementary cell fates?[J].EMBOReports,2014,15(11):1139-1153.
[3]CAMPISI J,D'ADDA D F F.Cellular senescence:when bad things happen to good cells[J].Nature Reviews Molecular Cell Biology,2007,8(9):729-740.
[4]CHILDS B G,DURIK M,BAKER D J,et al.Cellular senescence in aging and age-related disease:from mechanisms to therapy[J].Nature Medicine,2015,21(12):1424-1435.
[5]PAL S,TYLER J K.Epigenetics and aging[J].Science Advances,2016,2(7):e1600584.
[6]KENNEDY B K,BERGER S L,BRUNET A,et al.Geroscience:linking aging to chronic disease[J].Cell,2014,159(4):709-713.
[7]MOSKALEV A A,ALIPER A M,SMIT-MCBRIDE Z,et al.Genetics and epigenetics of aging and longevity[J].Cell Cycle,2014,13(7):1063-1077.
[8]CHANDRASEKARAN A,IDELCHIK M D P S,MELENDEZ JA.Redox control of senescence and age-related disease[J].Redox Biology,2017,11:91-102.
[9]VOSS T C,HAGER G L.Dynamic regulation of transcriptional states by chromatin and transcription factors[J].Nature Reviews Genetics,2014,15(2):69-81.
[10]BUENROSTRO J D,GIRESI P G,ZABA L C,et al.Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin,DNA-binding proteins and nucleosome position[J].Nature Methods,2013,10(12):1213-1218.
[11]LI B,CAREY M,WORKMAN J L.The role of chromatin during transcription[J].Cell,2007,128(4):707-719.
[12]RUIZ J L,TENA J J,BANCELLS C,et al.Characterization of the accessible genome in the human malaria parasite Plasmodium falciparum[J].Nucleic Acids Research,2018,46(18):9414-9431.
[13]TSOMPANA M,BUCK M J.Chromatin accessibility:a window into the genome[J].Epigenetics&Chromatin,2014,7:33.
[14]QUAN H,YANG Y,LIU S,et al.Chromatin structure changes during various processes from a DNA sequence view[J].Current Opinion in Structural Biology,2019,62:1-8.
[15]YU R,MCCAULEY B,DANG W.Loss of chromatin structural integrity is a source of stress during aging[J].Human Genetics,2020,139(3):371-380.
[16]BUENROSTRO J D,WU B,CHANG H Y,et al.ATAC-seq:a method for assaying chromatin accessibility genome-wide[J].Current Protocols in Molecular Biology,2015,109:21-29.
[17]CORCES M R,TREVINO A E,HAMILTON E G,et al.An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues[J].Nature Methods,2017,14(10):959-962.
[18]ZHENG R,WAN C,MEI S,et al.Cistrome Data Browser:expanded datasets and new tools for gene regulatory analysis[J].Nucleic Acids Research,2019,47(D1):D729-D735.
[19]FAN X,ZHU J,SCHADT E E,et al.Statistical power of phyloHMM for evolutionarily conserved element detection[J].Bio Med Central Bioinformatics,2007,8:374.
[20]SIEPEL A,BEJERANO G,PEDERSEN J S,et al.Evolutionarily conserved elements in vertebrate,insect,worm,and yeast genomes[J].Genome Research,2005,15(8):1034-1050.
[21]GU Q,YANG X,LV J,et al.AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate[J].Science,2019,363(6431):1085-1088.
[22]SCHWARTZ D M,FARLEY T K,RICHOZ N,et al.Retinoic acid receptor alpha represses a Th9 transcriptional and epigenomic program to reduce allergic pathology[J].Immunity,2019,50(1):106-120.e10.
[23]MILNER J J,TOMA C,YU B,et al.Runx3 programs CD8+Tcell residency in non-lymphoid tissues and tumors[J].Nature,2017,552(7684):253-257.
[24]GAO S,SONG Q,LIU J,et al.E2F1 mediates the downregulation of POLD1 in replicative senescence[J].Cellular and Molecular Life Sciences,2019,76(14):2833-2850.
[25]WANG J L,WANG P C.The effect of aging on the DNA damage and repair capacity in 2BS cells undergoing oxidative stress[J].Molecular Biology Reports,2012,39(1):233-241.
[26]SONG J,HONG P,LIU C,et al.Human POLD1 modulates cell cycle progression and DNA damage repair[J].Bio Med Central Biochemistry,2015,16:14.
[27]SHARPLESS N E,SHERR C J.Forging a signature of in vivo senescence[J].Nature Reviews Cancer,2015,15(7):397-408.
[28]MCHUGH D,GIL J.Senescence and aging:causes,consequences,and therapeutic avenues[J].The Journal of Cell Biology,2018,217(1):65-77.
[29]PARRY A J,HOARE M,BIHARY D,et al.NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence[J].Nature Communications,2018,9:1840.
[30]HE S,SHARPLESS N E.Senescence in health and disease[J].Cell,2017,169(6):1000-1011.





