基因组结构变异检测的基本方法与前沿技术
时间:2020-01-14 来源:基因组学与应用生物学 作者:杨金晶,李成,孙啸 本文字数:19296字摘 要: 本研究介绍了基因组结构变异检测的生物信息学基本方法和前沿技术。对基于第二代测序技术的四种检测方法 (读对方法, 读深方法, 分裂片段方法和序列拼接方法) 的原理和特点进行了详细解读, 分析了第二代测序技术应用在检测结构变异上的特点与发展趋势。最后介绍了三代测序、Linked-reads和光学物理图谱等新技术在基因组结构变异检测中的应用, 论述了融合新技术的结构变异检测方法的特点与优势。
关键词: 结构变异; 测序片段; 第二代测序技术; 长片段测序技术; 光学物理图谱技术;
Abstract: The basic methods and frontier technologies of genome structural variations detection were introduced in this paper. The principles and features of the 4 detection methods (Read-pair method, Read-depth method, Spiltread method and Sequence Assembly method) based on next generation sequencing technology were elaborated and the characteristics and development trend of the next generation sequencing technology on detecting structural variations were analyzed. Finally, some new technologies and their applications in detecting genome structural variations were introduced, including the third generation sequencing, linked-reads and optics physical maps. The features and advantages of the detection methods mixed with new technologies were discussed.
Keyword: Structural variations; Sequencing reads; Next generation sequencing; Long reads sequencing; Optics physical maps;
从基因的概念被提出伊始, 对人类自身基因信息的探究一直是生命科学的热门问题之一, 人类基因组计划 (human genome project, HGP) 于2001年第一次完成了人类24条染色体的序列测定后, 人们发现个体之间基因的相似程度达到99.9%, 存在着大约0.1%的片段上的差异, 我们称之为基因组的多态性或基因组变异, 正是这些差异导致了人与人之间截然不同的各类性状差异。
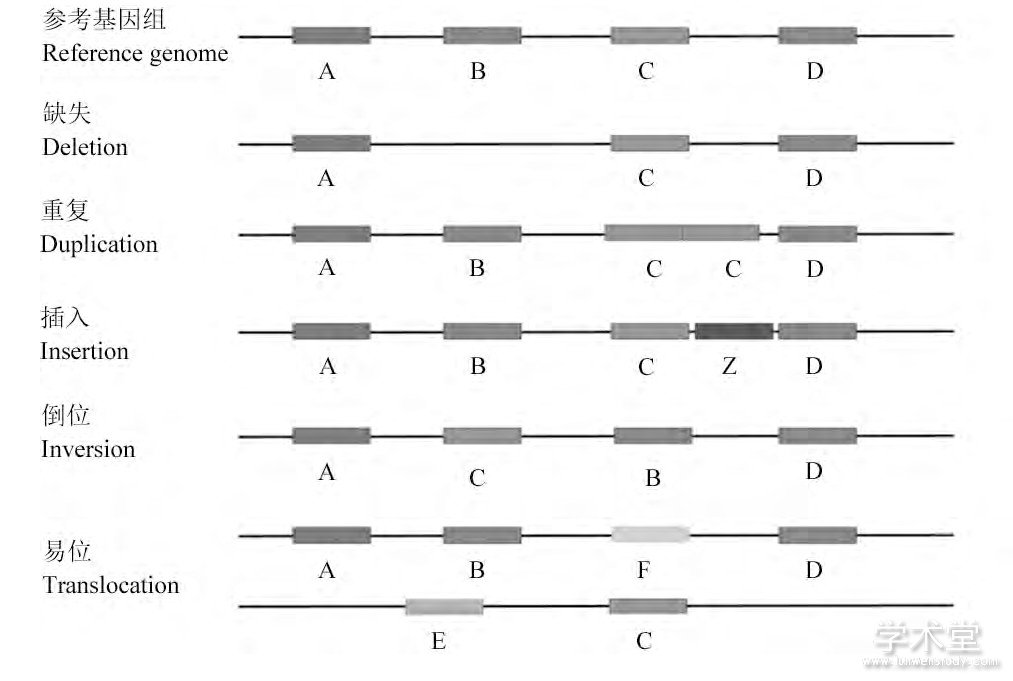
根据发生变异的碱基数量, 基因组变异又可以分为单核苷酸变异 (single nucleotide variations, SNV) 与结构变异 (structural variation, SV) 。SNV是指发生在基因组水平上的单个核苷酸的变异;SV最初提出是指长度在1 000 bp以上的基因的大片段的变异 (Feuk et al., 2006) , 随着对SV认识的不断发展, 现SV一般指长度在50 bp以上DNA片段变异 (Alkan et al., 2011) 。在结构变异中, 根据长度可以分为长度在3 MB以下的亚显微水平的结构变异和长度在3 MB以上的显微水平的结构变异;根据类型可以分为十多种不同的结构变异, 几种常见的类型为缺失 (Deletion) 、重复 (Duplication) 、插入 (Insertion) 、倒位 (Inversion) 、易位 (Translocation) 等 (图1) , 其中缺失、重复、插入等改变基因组碱基对数量的结构变异以及相互组合衍生出的复杂的结构变异又可以称为拷贝数变异 (copy number variation, CNV) (Cooper et al., 2007) 。
结构变异的影响可以归纳为两大方面 (Hurles et al., 2008) 。首先, 在基因表达方面, 结构变异会通过多种方式影响基因的转录与翻译。当基因发生重复、插入和缺失等变异时, 会导致基因剂量的改变;当编码区域发生结构变异时, 会改变基因的转录翻译;当非编码区域发生结构变异时, 会通过位置效应影响基因表达调控元件的调控作用;当发生增强子或抑制子的删除变异时, 会影响基因的转录水平。其次, 在疾病方面, 结构变异会导致性状的非正常表达, 从而引发各类遗传性疾病。除了已经为人们熟知的部分显微水平的结构变异引发的疾病, 例如21号染色体3体引发的唐氏综合征, 5号染色体短臂上的缺失引发的猫叫综合征等等;也有越来越多关于亚显微结构的结构变异引发的疾病的报道, 例如视蛋白基因的基因重组可能会引发红绿色盲疾病 (Lupski, 2015) ;17q21.31部位的缺失变异会引发学习障碍 (Koolen et al., 2006) ;16p11.2部位的缺失变异会引发孤独症 (W-eiss et al., 2008) 。

最初, 基因组中大量存在的SNV被认为是影响遗传和表型的主要因素, 但后来发现基因组中普遍存在大量的SV片段, 同样在人类疾病、复杂性状和进化的研究中具有重要意义 (Check, 2005) , 因此吸引了大量研究。一方面, 研究集中于人类基因组结构变异的检测。从2008年开始, 中、英、美各国共同发起的“国际千人基因组计划” (The 1000 Genomes Project) , 对基因组的结构变异作了当时最全面最完善的分析。在2012年和2015年, 国际千人基因组计划分别发布了1 092个样本 (Genomes Project et al., 2012) 和2 504个样本 (Sudmant et al., 2015) 的测序数据以及详细的结构变异检测结果。之后陆续有关于结构变异检测成果的报道, 到2016年10月, 韩国国立首尔大学医学院针对一名韩国人的基因组 (AK1) 进行了相关分析 (Seo et al., 2016) , 发布了迄今为止最为详细的人类基因组结构变异检测结果。另一方面, 人们关注于结构变异与相关疾病的关联分析, 已经有多种自身免疫性疾病 (Yang et al., 2007;Wang et al., 2013) 、病毒感染 (Gonzalez et al., 2005) 、肥胖 (Falchi et al., 2014) 、骨质疏松 (Yang et al., 2008) 等被证明与结构变异相关, 尤其在癌症与结构变异的关联性研究中, 更是发现结构变异是导致食道癌 (Cheng et al., 2016) 、儿童神经母细胞瘤 (Pugh et al., 2013) 、小细胞肺癌 (George et al., 2015) 等最主要的因素。
图1 结构变异的几种常见类型Figure 1 Several common types of structural variations
其实早在上世纪五十年代, 对于结构变异的研究便已经开始, 但受限于技术手段, 过去人们往往只能通过显微镜观察到显微水平的结构变异。上世纪七十年代, 人们用遗传学方法对结构变异进行了更深入的研究 (Sperling and Wiesner, 1972) 。21世纪以来, 一方面随着微阵列 (Microarrays) 、细菌人工染色体 (bacteria artificial chromosome, BAC) 、单分子分析 (Single-molecule analysis) 等实验技术的发展, 人们开始使用阵列比较基因组杂交 (array comparative genomic hybridization, aCGH) 、SNP微阵列 (SNP microarrays) 以及荧光原位杂交 (fluorescent in situ hybridization, FISH) 等方法来检测结构变异 (Iafrate et al., 2004) 。另一方面, 随着聚合酶链式反应 (polymerase chain reaction, PCR) 、DNA测序以及基因组序列比较分析等技术的发展, 人们开始通过基于测序数据的计算机处理方法检测结构变异, 尤其随着新一代测序技术 (next generation sequencing, NGS) 的发展和普及, 基于测序数据的分析方法开始被大量使用。近几年来, 为了弥补NGS技术检测结构变异的各种不足, 人们开始通过单分子实时测序 (single-molecule realtime sequencing, SMRT) 、纳米孔 (Nanopore) 等第三代测序技术 (third generation sequencing, TGS) 进行SV检测。本研究主要就基于测序技术发展起来的一系列检测结构变异的方法和技术进行介绍和讨论。
1、 基因组结构变异检测基本方法
每段DNA的测序序列的原始数据称之为测序片段 (Reads) , 基于测序技术的结构变异检测方法大部分通过reads与参考基因组的比对进行检测。主要检测方法分为四种 (Medvedev et al., 2009;Alkan et al., 2011;Mills et al., 2011) , 分别是读对方法 (Read-pair Method) 、读深方法 (Read-depth method) 、分裂片段方法 (Split-read method) 以及序列拼接方法 (Sequence assembly method) 。
1.1、 读对方法
将同一段DNA分别从两端测得不同方向的序列信息称之为双端测序 (Paired-end reads) 。读对方法通过双端测序, 获得DNA片段两端成对reads的分布的信息, 再寻找比对到参考基因组上后分布和方向与参考基因组不一致的Reads, 以此为特征判断结构变异的类型 (Alkan et al., 2011) 。
读对方法以PEM算法 (Korbel et al., 2007) 、BreakDancer算法 (Chen et al., 2009) 、HYDRA算法 (Quinlan et al., 2010) 等为代表。以PEM算法为例, 首先对样本DNA进行双端测序 (图2A) , 可以获得DNA片段两端成对reads的距离和方向等信息。之后将测得的成对的reads比对到参考基因组上, 分析其在参考基因组上的距离和方向信息, 根据比对前后距离和方向信息的不一致性 (图2B) , 来判断是否存在SV。发生缺失变异的片段两端的reads在比对到参考基因组上时, 其距离会增大, 而发生插入变异的片段则会出现距离减少的情况, 发生倒位变异的片段会出现方向上的变化。
读对方法是基于高通量测序数据检测结构变异的方法中使用最广泛的, 最早通过乳腺癌细胞系MCF-7产生的BAC序列验证该方法的可行性 (Volik et al., 2003) 。理论上读对方法可以检测各种类型的结构变异, 但是在处理基因组重复区域的比对时会受到很大干扰。同时因为DNA片段长度的限制, 读对方法无法检测大片段的结构变异。
1.2、 读深方法
读深方法首先假设在参考基因组上测序深度 (Read depth) 是随机分布的 (通常服从泊松分布或者修正泊松分布) 。将通过高通量测序获得的样本基因组的reads比对参考基因组上, 分析其测序深度, 通过测序深度在某些区域的差异变化来发现重复变异和缺失变异:重复区域的测序深度会出现明显增加, 缺失区域的测序深度会出现明显减少 (Alkan et al., 2011) 。
读深方法以EWT算法 (Yoon et al., 2009) 、CNV-nator算法 (Abyzov et al., 2011) 等为代表。以EWT算法为例, 首先在参考基因组上每100 bp取互不重叠的窗, 计算每个窗中比对到参考基因组上的reads的起始位点的个数 (图3A) , 再乘以与基因组中GC含量相关的比例系数, 作为每个窗的序列深度。依次计算每个窗中的测序深度, DNA片段上所有窗的测序深度总体应当近似服从泊松分布, 但如果出现缺失变异、重复变异等拷贝数变异, 则必然会引起连续的窗中的序列深度发生明显的增加或降低的情况 (图3B) 。
图2 PEM算法检测SV的流程与特征Figure 2 The workflow and features of PEM algorithm for SVdetection
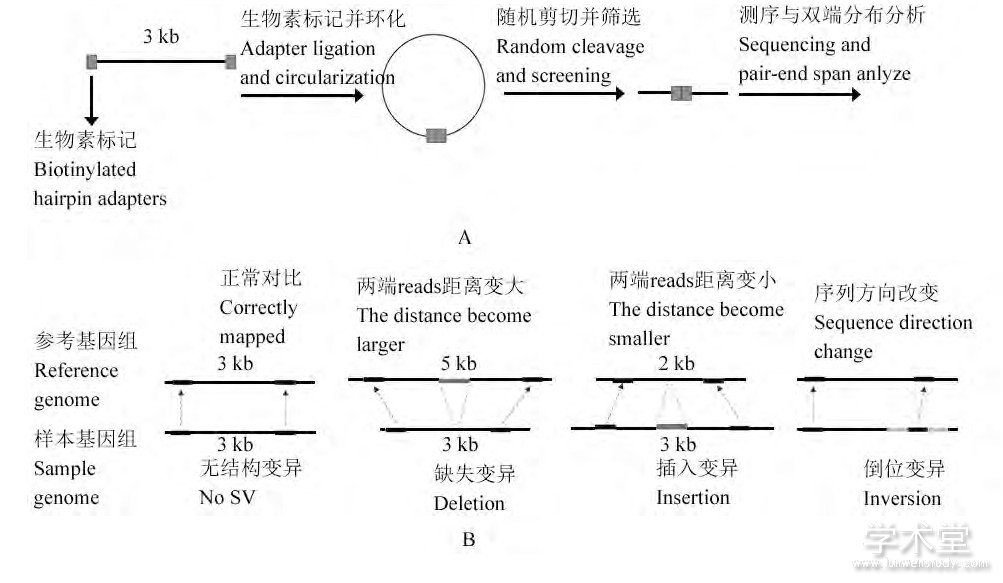
注:A:双端测序过程, 将基因组DNA剪切成长度为3 kb左右的DNA片段, 在片段两端用生物素标记后环化, 再将环化片段随机剪切, 筛选出具有生物素标记的片段, 然后对筛选出的片段进行测序, 进而分析获得DNA片段两端成对reads的距离和方向信息;B:不同结构变异检测时的不同特征, 假设原本DNA片段长度为3 kb, 两端序列在比对到参考基因组上后, 若距离变为了2 kb, 则DNA片段中可能出现了插入变异;若距离变成了5 kb, 则可能出现了缺失变异;若一端的序列出现方向上的变化, 则可能出现倒位变异Note:A:The figure of progress of paired-end sequencing.The genome DNA was sheared to yield DNA fragments of 3 kb, and then the fragments were labeled by biotin at both ends and circularized.And the circularized fragments were randomly sheared and the biotinylated fragments were screened, then the selected fragments were sequenced, and the distance and direction information of the pair-end reads of the DNA fragments were obtained;B:The figure of various features when detecting different kinds of structural variations;Suppose that the length of the original DNA fragments is 3 kb.If the length becomes 2 kb after their paired-end reads are mapped to the reference genome, there might be insertions in the DNA fragment;if the length becomes 5 kb, there might be deletions;if one of the reads'direction changes, there might be inversions
读深方法是通过reads比对的统计信息检测结构变异的方法, 其最早被用来解释在癌症细胞中发生的基因重组的现象 (Campbell et al., 2008) 。读深方法在检测基因组重复、缺失结构变异时的效果非常显着, 且可以用来预测基因的拷贝数, 但其无法检测其他类型的结构变异, 无法区分串联重复和散在重复, 而且读深方法无法获得断点的相关信息, 只能判断片段中是否存在结构变异, 而不能判断出结构变异的准确位置。
图3 EWT算法检测SV的原理Figure 3 The principle of EWT algorithm for SV detection
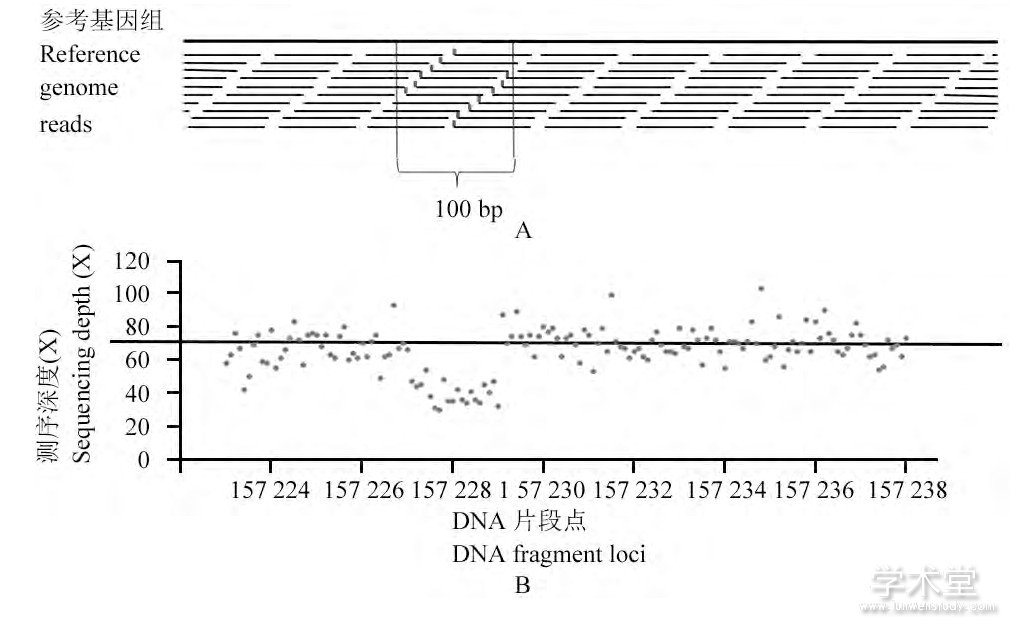
注:A:EWT算法计算测序深度过程;方框的长度为100 bp, 以此作为一个窗, 计算窗内reads的起始位点 (标记区域内) 个数, 作为这个窗的测序深度的计算标准;B:模拟的缺失变异样本基因组的测序深度分布情况;在样本157 224~157 238 kb的长度为14 kb的DNA片段上共构建了140个窗, 这些窗的测序深度的分布在正常情况下近似服从期望为70的泊松分布;在157 227~157 229 kb的区域内, 序列深度出现了连续且明显的降低则可以判断在这一区域内出现了缺失变异Note:A:The progress of calculating read-depth in EWT algorithm;The length of the box is 100 bp, it is called a window, the number of start points (the marked region) of the reads in this window is the standard of the read-depth;B:The distribution of read-depths of simulated sample genome with deletions;From the point 15 722 kb to the point 157 238 kb, the DNA fragment's length is 14kb;We built 140 windows, the distribution of the windows ought to obey the poisson distribution whose expectation is 70;In the region be tween 157 227 kb and 157 229 kb, the read-depth decreased obvi ously and continuously;It can be judged that here might be deletions in this region
1.3、 分裂片段方法
样本基因组测序获得的reads通常要比对到参考基因组上, 由于发生结构变异, 在某些reads的某个位置的左右两侧, 碱基对的坐标和方向与参考基因组不一致, 这个位置被称为断点 (Break point) 。分裂片段方法通过寻找结构变异样本中含有断点的reads上准确的断点位置信息来检测结构变异 (Alkan et al., 2011) (图4A) 。分裂片段方法将样本基因的各个reads比对到参考基因组上, 寻找无法比对的reads, 分别在无法比对的reads的特定碱基位置设置断点, 按断点分裂成两小段reads, 再通过观察两个小段reads比对到参考基因组中的情况, 从而判断结构变异情况。
分裂片段方法以Pindel算法 (Ye et al., 2009) 、AGE算法 (Abyzov and Gerstein, 2011) 等为代表。以Pindel算法为例, 首先通过SSAHA2软件将所的reads比对到参考基因组上, 寻找其中一端能比对到基因组上而另一端无法比对的reads, 再从可以比对的一端开始使用模式增长 (Pattern growth) 算法搜索最大-最小子串, 来寻找断点的精确位置, 再将reads按断点分裂成两段, 将片段分别比对到基因组上, 来判断结构变异的具体信息 (图4B) 。
分裂片段方法基于对reads的分段来检测结构变异的断点, 可以检测单碱基分辨率的缺失变异和插入变异, 对有明确的断点特征的结构变异具有很好的检测效果, 当reads的长度大于插入片段的长度时, 分裂片段方法的拓展还可以用来检测移动元素插入 (mobile-element insertions, MEI) (Mills et al., 2011) 。但仍有大量的结构变异不存在断点特征, 无法通过分裂片段方法检测, 且其在具有大量重复片段的区域检测效果不佳。分裂片段方法最早是基于Sanger测序法开发的 (Mills et al., 2006) , 测序片段越长, 检测效果越好, 二代测序数据读长短的特点会严重影响分裂片段方法检测的效果。
1.4、 序列拼接方法
序列拼接方法通过对样本基因组的reads片段进行从头拼接 (De novo assembly) , 重新组装后解码样本基因组的序列, 再将其与参考基因组序列进行比对, 从而可以清楚地判断是否存在结构变异以及结构变异类型 (Alkan et al., 2011) 。
序列拼接的方法以ABy SS算法 (Simpson et al., 2009) 、Velvet算法 (Zerbino and Birney, 2008) 和SOA-Pdenovo算法 (Li et al., 2010) 等为代表。以ABy SS算法为例, 首先根据目标k值, 通过测序片段产生所有可能的长度为k的子串, 移除子串数据集读取误差, 再通过de Bruijn图算法构建初始的重叠群 (Contigs) , 之后使用配对信息来消除Contigs的重叠模糊性, 拓展Contigs的范围, 从而获得最后的拼接结果 (图5A) 。用拼接获得的完整的样本基因组片段与参考基因组片段进行比对时, 在未发生结构变异的区域比对完全一致, 在发生结构变异的区域比对则会出现差异 (图5B) 。
相对于前三种方法, 序列拼接方法采用了截然不同的非reads比对的思路。从理论上来说, 如果能够拼接样本基因组的全部序列, 则可以检测出所有的SV与SNV, 但以测序长度为100 bp的Illumina测序仪为代表的第二代测序技术普遍读长偏短, 使得拼接难度大大提升, 同时如果基因组上出现大量重复片段时, 会引发拼接算法的崩溃性错误 (Chaisson et al., 2015) 。如何提高测序片段长度并改进序列拼接的算法是序列拼接方法亟待解决的问题。
图4 使用分裂片段方法检测SV原理Figure 4 The principle of Split-read method for SV detection
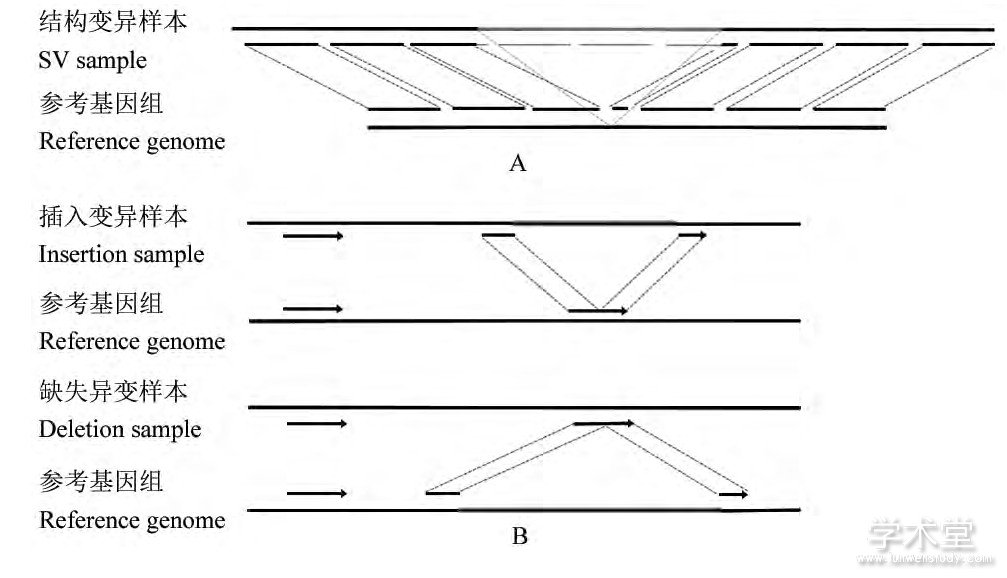
注:A:结构变异的断点示意, 样本基因组标记区域内为缺失变异区域, 在结构变异区域之外的reads可以正确比对到参考基因组上, 结构变异区域的reads无法正确比对到参考基因组上, 在结构变异区域的起始和终止位置的reads, 其标记之外的部分是可以正确比对的, 标记处的位置即为reads的断点;B:不同结构变异检测时的不同特征, 发生插入变异的DNA片段, 插入片段前后的reads在断点处各有一部分可以比对到参考基因组上的相邻位置;发生缺失变异的DNA片段, 缺失部分的reads按断点可以分别比对到参考基因组前后不同位置Note:A:The figure of the break point of SV, the region inside the marked area is deletion region, the Reads out of the mark can be mapped to the reference genome correctly, and the Reads in the variation regions cannot be mapped to the reference genome;In the start and end regions, the part out of the mark can be correctly mapped, and the mark positions are the break points of the Reads;B;The figure of various features when detecting different kinds of structural variants.In the DNA fragments with insertions, the Reads before and after the insertion region can be partly mapped to the adjacent positions of the reference genome;in the DNA fragments with deletions, the reads can be partly mapped to dispersed positions in reference genome
1.5、 当前结构变异检测方法的特点及发展趋势
目前的测序技术以第二代测序技术为主, 第二代测序技术又称为新一代测序技术, NGS技术的代表是Illumina公司的测序仪, 其每次产生的reads长度在100 bp左右, 重要特点是技术成熟、通量高、测序成本低、测序速度快, 是目前基因组测序的主要手段。借助NGS技术, 可以通过单次测序实验发现不同类型的结构变异, 而且得益于NGS技术的高准确度, 可以准确检测出基因组的拷贝数变化, 且具备了发现完整基因组变异的潜力。同时由于NGS技术高通量的特点, 提高了结构变异检测效率并降低了其成本。但是, NGS技术存在读长短的缺陷, 会制约读对和分段方法的检测效率, 且对序列拼接方法带来极大困难。使用NGS数据检测结构变异的灵敏度不高, 且大多局限于短片段的缺失变异和插入变异, 无法检测大片段的复杂结构变异。
图5 序列拼接方法检测SV原理Figure 5 The principle of Sequence assembly method for SV detection
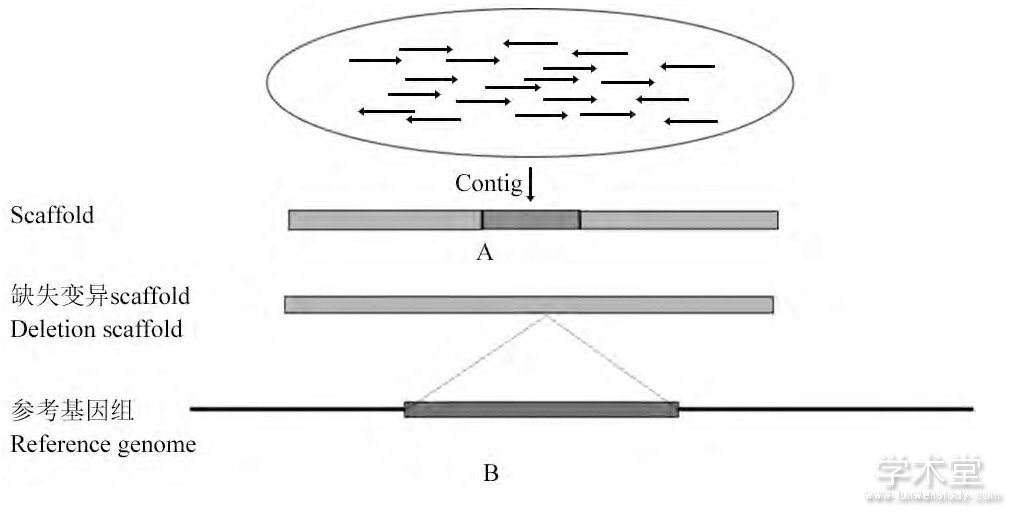
注:A:为序列拼接过程, 通过大量相互重叠的reads进行拼接, 可以获得长度较长的Contigs, 再对Contigs进行拼接, 可以获得长片段Scaffold;B部分为含有缺失变异的Scaffold与参考基因组比对示意图, 非缺失部分的序列都可以正常比对到基因组上, 缺失部分则无法正常比对, 由此可以非常直观地得到变异区域的具体信息Note:A:The figure of the progress of sequence assembly, The long Contigs can be achieved by assembling large number of overlapping reads, and the Scaffold can be achieved by assembling contigs;B:The figure of the result the Scaffold with deletions mapped to the reference genome;The normal part can be mapped to the reference genome correctly, but the deletion part cannot be mapped correctly;According to this, the specific information of the variant parts can be obtained directly
在国际千人基因组计划于2012年发布1 092个个体的结构变异检测结果中, 所有样本的数据均通过低覆盖度NGS获得, 包括6 x覆盖度的全基因组测序 (whole-genome sequencing, WGS) 和全外显子组测序 (whole-exome sequencing, WES) , 运用BreakDancer、CNVnator、Delly、Pindel、Genome STRiP (Handsaker et al., 2011) 等结构变异检测算法, 检测了14 000多个大片段的缺失变异以及小片段的串联重复序列;而在2015年发布的结构变异检测结果中, 除了使用了低覆盖度的全基因组测序, 还加入了单分子实时测序、SNP微阵列等各种技术相结合的测序手段, 使用同样的算法, 共检测了68 000多个结构变异, 包括了缺失、重复、倒置、插入等不同类型的结构变异, 其中有48 000多个结构变异是从未发现的, 而且近一半的结构变异没有明显的断点特征。
对比来看, 由于测序技术的区别, 虽然采用相同的算法, 但两次检测结构变异的结果存在巨大差异。仅仅采用低覆盖度的二代测序数据只能检测出相对少量的SV, 且大多只局限于缺失变异。同时, 不同的结构变异被检测出的程度也不尽相同, 据估计, 68%的倒位变异和35%的重复变异尚未被检测出;相反, 80%的缺失变异已经被检测。所以, 仅仅采用低覆盖度的二代测序产生的数据来检测结构变异已经逐渐无法满足检测的需求。
对于如何提高结构变异的检测水平, 可以从3个方面入手 (Huddleston and Eichler, 2016) 。 (1) 提高测序深度, 改进测序形式:NGS的测序深度至少要达到30 x, 而不是简单的6×覆盖度, 这样才可以提高检测结构变异的灵敏度。同时最好以家庭为单位来进行测序, 以了解表型特征的传递以及变异频率等信息; (2) 提高测序长度, 完善序列拼接算法:使用单分子实时测序 (Single molecule real-time sequencing) 等长片段测序方法提高Reads长度, 随着读长增加, 序列拼接算法的效果会出现显着提高, 序列拼接的难度也会显着降低, 实现基因组的完全解码成为可能; (3) 综合使用检测算法, 采用读深方法、读对方法、分裂片段方法和序列拼接方法相结合的结构变异检测方法, 例如CNVer算法 (Medvedev et al., 2010) 、Genome STRiP算法等弥补单一方法的不足。
2、 结构变异检测前沿技术和新方法
2.1、 基因组分析新技术
近几年来, 在基因组分析上出现了许多新技术, 这些技术都围绕着获取长片段的基因组测序序列的进行, 主要分为三类: (1) 直接获取长片段的新测序技术, 即第三代测序技术; (2) 对NGS获得的短片段进行处理获取长片段的技术, 即连Link-reads技术; (3) 构建基因组物理图谱辅助序列拼接的技术, 即光学图谱技术。
第三代测序技术以Pacific BioSciences公司的单分子实时测序 (single molecule real-time, SMRT) 技术 (Rhoads and Au, 2015) 为代表。SMRT技术通过荧光信号获取序列信息, 其优点是读长超长, 平均读取长度可以达到16 kb左右, 在基因组组装和结构变异检测方面可以起突破性的作用。然而三代测序技术相较于二代测序技术错误率高, 准确率在85%左右, 虽然可以通过多次重复测序使测序准确率达到95%以上, 但成本也会成倍增加;测序通量低, 单次测序的通量是MB级别, 与NGS的通量差距巨大, 因此测序成本高, 无法大规模应用。
Linked-reads技术 (Kitzman, 2016) 以10X Genomics公司的GemCode平台为代表。GemCode平台对基因组上同一区域内的DNA片段标记以相同的特殊碱基序列, 在通过Illumina平台测序后, 连接相同特殊碱基序列标记的DNA片段, 产生一种新的数据类型:连接片段 (Linked-reads) , 从而可以以相对较低的成本来获得长度达到10 kb以上的测序片段, 进而能更好地进行基因组组装并提高结构变异检测灵敏度。Gemcode的缺点在于其对样本质量要求高, 需要制备大小不同的文库, 且其测序基础是基于Illumina测序的, 所以无法改善高GC或低GC含量时测序覆盖效果较差的情况 (Ross et al., 2013) 。
光学图谱技术又被称为新一代图谱 (next-generational mapping, NGM) 技术, 以BioNano公司的Irys平台为代表。Irys平台通过酶切技术和荧光标记成像技术构建基因组的物理图谱, 描绘DNA上可以识别的标记的位置 (包括限制性内切酶的酶切位点, 基因等) 和相互之间的距离, 构建基因组的宏观框架, 依照框架可以使测序信息准确地回归到染色体上, 从而提高序列拼接的长度和准确度, 解决在高度重复区域的基因组组装和结构变异检测问题。在基因组分析方面, 光学图谱技术只是一项辅助技术, 但其能够很好地还原DNA分子的真实信息, 辅助序列重新组装, 并且能够与第二、第三代测序技术完美兼容, 具有重要的应用价值。
2.2、 融合长片段测序和物理图谱的结构变异检测方法
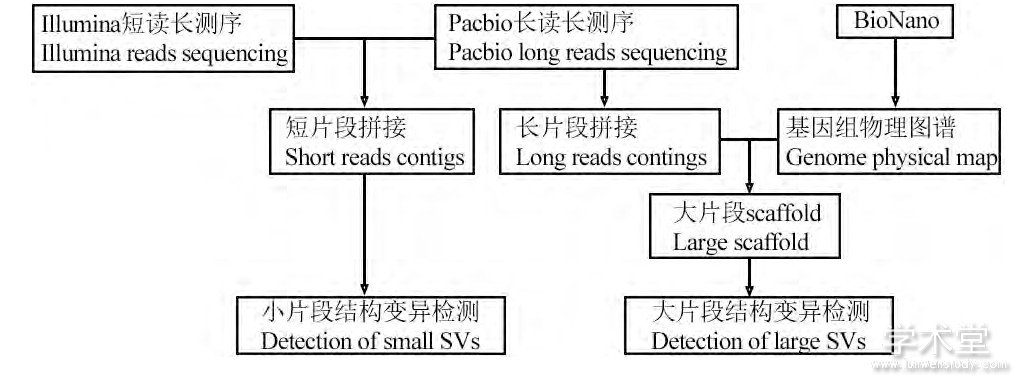
随着上述新技术的出现, 基因组测序的片段长度大大提高, 弥补了序列拼接方法的缺陷, 其检测效果获得了突破性地提高, 可以检测大片段和复杂的结构变异。从最新的关于结构变异的相关报道来看, 以NGS短片段数据结合长片段测序数据, 辅助以基因组物理图谱技术, 使用序列拼接方法检测结构变异的流程大概分为两个部分 (图6) : (1) 对长测序片段进行序列拼接, 形成长度在MB级别的Contigs, NGS短片段补充细节, 将Bionano基因组图谱与Contigs相结合, 构建大片段的Scaffolds, 与参考基因组比对, 检测结构变异; (2) 以长片段数据为框架, 对NGS短片段数据进行序列拼接, 将拼接获得的Contigs与参考基因组比对, 检测结构变异。基于以上方法, 在结构变异的检测上有了新的突破。
2015年6月, Pacific Biosciences (PacBio) 公司给出了SMRT测序组装人类基因组的成果 (Pendleton et al., 2015) , 选用的样本是NA12878。其主要使用SMRT测序数据结合Bionano物理图谱技术, 构建样本基因组Scaffolds, 再使用NGS测序数据填补缺口, 使用序列拼接方法等进行结构变异检测。使用SMRT测序数据拼接获得的Contigs的N50长度可以达到900 kb以上, Scaffold的N50长度高达30 MB, 相对于NGS测序数据拼接的长度有了显着提高。在检测结构变异方面, 除了检测出了各种小片段的结构变异, 以及类型为插入、缺失以及片段重复的90多个长度在6 kb以上的长片段SV, 更是通过基因组图谱检测出了长度在100~400 kb之间的8个大片段缺失变异与11个大片段插入变异。
2016年12月, 10×Genomics公司给出了Linkedreads测序组装人类基因组的结果 (Mostovoy et al., 2016) , 选用的样本同样是NA12878。其首先对NGS数据使用SOAP de novo算法进行拼接, 再结合10×Genomics的Gemcode平台产生的连接读取数据, 形成大片段的scaffold, 最后再与Bionano物理图谱相结合, 产生最后的序列拼接结果, 与参考基因组进行比对, 检测结构变异。最终综合各种方法拼接的scaffold的N50长度同样可以达到与SMRT技术相同的30 MB以上。在检测结构变异方面, 该实验同样检测出了各种小片段的插入、删除变异, 同时还给出了200个大片段的重复变异的具体分布情况。
2016年韩国国立首尔大学医学院给出的AK1基因组的相关分析中, 综合PacBio长读长测序, Illumina短读长测序, 10×Genomics连接片段, BioNano Genomics光学图谱以及细菌人工染色体 (BAC) 等方法, 对AK1基因组进行从头组装和基因组分析, 在亚洲人的基因组结构变异检测方面取得了大量进展。其首先使用Illumina短读长测序数据, 结合10×G的Gemcode与BAC产生的数据, 进行序列拼接, 检测结构变异;再使用PacBio长读长测序数据进行序列拼接, 结合Bionano物理图谱技术, 构建Scaffold, 检测长片段结构变异。通过PacBio测序数据拼接获得的Contigs的N50长度可以达到17.7 MB, 而最终综合拼接获得的Scaffold的N50长度达到了44.8 MB。在检测结构变异方面共鉴定到了18 210个大片段的结构变异, 包含7 358个缺失, 10 077个插入, 71个倒置和704个复杂变异, 其中47%的缺失变异与76%的插入变异等都是未曾报道过的。
图6 融合NGS, 长片段测序与物理图谱的结构变异检测方法流程Figure 6 The workflow of SV detection combined NGS, long reads sequencing and physical maps
3、 总结与思考
随着测序技术的迅猛发展以及基因组分析技术的不断进步, 人们对人类基因组的结构变异逐步有了详细与系统的认识。基于测序技术的读深、读对、分裂片段、序列拼接方法为结构变异的检测提供了高效准确的方法, 开拓了实验与计算机数据处理相结合检测结构变异的新模式, 即使各项技术不断进步, 依然在围绕着这些方法展开。
随着新技术的发展, 测序片段的长度不断增加, 检测结构变异的准确度和灵敏度也在不断提高, 而从最新的一些报道来看, 无一例外的都选择了序列拼接方法主导的检测方法。一方面, 虽然四种基本方法在基于短片段的结构变异检测上都有各自不可替代的优势, 但序列拼接方法通过解码基因组序列, 能够更加直观、直接地检测所有类型的结构变异, 并可以精准判断出不同长度、不同类型的结构变异的具体位置。随着测序片段长度达到10 kb甚至更长后, 序列拼接方法的准确性大大提高, 准确拼接长片段的技术难度也随之降低。另一方面, 随着序列拼接方法逐渐成为结构变异的主流检测方法, 对序列拼接方法相关算法的研究也在不断深入, 更多高效高准确率的算法在不断提出, 序列拼接方法在不断彰显其蓬勃的生命力。
作者贡献
杨金晶负责论文的整体框架设计, 文献资料总结以及文稿写作, 李成负责文献资料补充和综述文稿的修改, 孙啸是论文的指导者及负责人, 指导论文架构设计, 论文写作与修改。全体作者都阅读并同意最终的文本。
参考文献
[]Abyzov A., and Gerstein M., 2011, AGE:defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision, Bioinformatics, 27 (5) :595-603
[]Abyzov A., Urban A.E., Snyder M., and Gerstein M., 2011, CN-Vnator:an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing, Genome Research, 21 (6) :974-984
[]Alkan C., Coe B.P., and Eichler E.E., 2011, Genome structural variation discovery and genotyping, Nature Reviews Genetics, 12 (5) :363-376
[]Campbell P.J., Stephens P.J., Pleasance E.D., O'Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C., Teague J.W., Menzies A., Goodhead I., Turner D.J., Clee C.M., Quail M.A., Cox A., Brown C., Durbin R., Hurles M.E., Edwards P.A.W., Bignell G.R., Stratton M.R., and Futreal P.A., 2008, Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel pairedend sequencing, Nature Genetics, 40 (6) :722-729
[]Chaisson M.J., Wilson R.K., and Eichler E.E., 2015, Genetic variation and the de novo assembly of human genomes, Nature Reviews Genetics, 16 (11) :627-640
[]Check E., 2005, Human genome:patchwork people, Nature, 437 (7062) :1084-1086
[]Chen K., Wallis J.W., McLellan M.D., Larson D.E., Kalicki J.M., Pohl C.S., McGrath S.D., Wendl M.C., Zhang Q., Locke D.P., Shi X., Fulton R.S., Ley T.J., Wilson R.K., Ding L., and Mardis E.R., 2009, Break dancer:an algorithm for high-resolution mapping of genomic structural variation, Nature Methods, 6 (9) :677-681
[]Cheng C., Zhou Y., Li H., Xiong T., Li S., Bi Y., Kong P., Wang F., Cui H., Li Y., Fang X., Yan T., Li Y., Wang J., Yang B., Zhang L., Jia Z., Song B., Hu X., Yang J., Qiu H., Zhang G., Liu J., Xu E., Shi R., Zhang Y., Liu H., He C., Zhao Z., Qian Y., Rong R., Han Z., Zhang Y., Luo W., Wang, J., Peng S., Yang X., Li X., Li L., Fang H., Liu X., Ma L., Chen Y., Guo S., Chen X., Xi Y., Li G., Liang J., Yang X., Guo J., Jia J., Li Q., Cheng X., Zhan Q., and Cui Y., 2016, Whole-genome sequencing reveals diverse models of structural variations in esophageal squamous cell carcinoma, American Journal of Human Genetics, 98 (2) :256-274
[]Cooper G.M., Nickerson D.A., and Eichler E.E., 2007, Mutational and selective effects on copy-number variants in the human genome, Nature Genetics, 39 (7S) :22-29
[]Falchi M., El-Sayed Moustafa J.S., Takousis P., Pesce F., Bonnefond A., Andersson-Assarsson J.C., Sudmant P.H., Dorajoo R., Al-Shafai M.N., Bottolo L., Ozdemir E., So H.C., Davies R.W., Patrice A., Dent R., Mangino M., Hysi P.G., Dechaume A., Huyvaert M., Skinner J., Pigeyre M., Caiazzo R., Raverdy V., Vaillant E., Field S., Balkau B., Marre M., Visvikis-Siest S., Weill J., Poulain-Godefroy O., Jacobson P., Sjostrom L., Hammond C.J., Deloukas P., Sham P.C., McPherson R., Lee J., Tai E.S., Sladek R., Carlsson L.M., Walley A., Eichler E.E., Pattou F., Spector T.D., and Froguel P., 2014, Low copy number of the salivary amylase gene predisposes to obesity, Nature Genetics, 46 (5) :492-497
[]Feuk L., Carson A.R., and Scherer S.W., 2006, Structural variation in the human genome, Nature Reviews, Genetics, 7 (2) :85-97
[]Genomes Project C., Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., and McVean G.A., 2012, An integrated map of genetic variation from 1 092 human genomes, Nature, 491 (7422) :56-65
[]George J., Lim J.S., Jang S.J., Cun Y., Ozretic L., Kong G., Leenders F., Lu X., Fernandez-Cuesta L., Bosco G., Muller C., Dahmen I., Jahchan N.S., Park K.S., Yang D., Karnezis A.N., Vaka D., Torres A., Wang M.S., Korbel J.O., Menon R., Chun S.M., Kim D., Wilkerson M., Hayes N., Engelmann D., Putzer B., Bos M., Michels S., Vlasic I., Seidel D., Pinther B., Schaub P., Becker C., Altmuller J., Yokota J., Kohno T., I-wakawa R., Tsuta K., Noguchi M., Muley T., Hoffmann H., Schnabel P.A., Petersen I., Chen Y., Soltermann A., Tischler V., Choi C.M., Kim Y.H., Massion P.P., Zou Y., Jovanovic D., Kontic M., Wright G.M., Russell P.A., Solomon B., Koch I., Lindner M., Muscarella L.A., la Torre A., Field J.K., Jakopovic M., Knezevic J., Castanos-Velez E., Roz L., Pastorino U., Brustugun O.T., Lund-Iversen M., Thunnissen E., Kohler J., Schuler M., Botling J., Sandelin M., Sanchez-Cespedes M., Salvesen H.B., Achter V., Lang U., Bogus M., Schneider P.M., Zander T., Ansen S., Hallek M., Wolf J., Vingron M., Yatabe Y., Travis W.D., Nurnberg P., Reinhardt C., Perner S., Heukamp L., Buttner R., Haas S.A., Brambilla E., Peifer M., Sage J., and Thomas R.K., 2015, Comprehensive genomic profiles of small cell lung cancer, Nature, 524 (7563) :47-53
[]Gonzalez E., Kulkarni H., Bolivar H., Mangano A., Sanchez R., Catano G., Nibbs R.J., Freedma B.I., Quinones M.P., Bam shad M.J., Murthy K.K., Rovin B.H., Bradley W., Clark R.A., Anderson S.A., O'Connell R.J., Agan B.K., Ahuja S.S., Bologna R., Sen L., Dolan M.J., and Ahuja S.K., 2005, The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility, Science, 307 (5714) :1434-1440
[]Handsaker R.E., Korn J.M., Nemesh J., and Mc Carroll S.A., 2011, Discovery and genotyping of genome structural polymorphism by sequencing on a population scale, Nature Genetics, 43 (3) :269-276
[]Huddleston J., and Eichler E.E., 2016, An incomplete understanding of human genetic variation, Genetics, 202 (4) :1251-1254
[]Hurles M.E., Dermitzakis E.T., and Tyler-Smith C., 2008, The functional impact of structural variation in humans, Trends in Genetics Tig, 24 (5) :238-245
[]Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., and Lee C., 2004, Detection of largescale variation in the human genome, Nature Genetics, 36 (9) :949-951
[]Kitzman J.O., 2016, Haplotypes drop by drop, Nature Biotechnology, 34 (3) :296-298
[]Koolen D.A., Vissers L.E., Pfundt R., de Leeuw N., Knight S.J., Regan R., Kooy R.F., Reyniers E., Romano C., Fichera M., Schinzel A., Baumer A., Anderlid B.M., Schoumans J., Knoers N.V., van Kessel A.G., Sistermans E.A., Veltman J.A., Brunner H.G., and de Vries B.B., 2006, A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism, Nature Genetics, 38 (9) :999-1001
[]Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L., Taillon B.E., Chen Z., Tanzer A., Saunders A.C., Chi J., Yang F., Carter N.P., Hurles M.E., Weissman S.M., Harkins T.T., Gerstein M.B., Egholm M., and Snyder M., 2007, Paired-end mapping reveals extensive structural variation in the human genome, Science, 318 (5849) :420-426
[]Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K., Li S., Yang H., Wang J., and Wang J., 2010, De novo assembly of human genomes with massively parallel short read sequencing, Genome Research, 20 (2) :265-272
[]Lupski J.R., 2015, Structural variation mutagenesis of the human genome:Impact on disease and evolution, Environmental and Molecular Mutagenesis, 56 (5) :419-436
[]Mostovoy Y., Levy-Sakin M., Lam J., Lam E.T., Hastie A.R., Marks P., Lee J., Chu C., Lin C., Dzakula Z., Cao H., Schlebusch S.A., Giorda K., Schnall-Levin M., Wall J.D., and Kwok P.Y., 2016, A hybrid approach for de novo human genome sequence assembly and phasing, Nature Methods, 13 (7) :587-590
[]Medvedev P., Fiume M., Dzamba M., Smith T., and Brudno M., 2010, Detecting copy number variation with mated short reads, Genome Research, 20 (11) :1613-1622
[]Medvedev P., Stanciu M., and Brudno M., 2009, Computational methods for discovering structural variation with next-generation sequencing, Nature Methods, 6 (11 Suppl) :S13-S20
[]Mills R.E., Luttig C.T., Larkins C.E., Beauchamp A., Tsui C., Pittard W.S., and Devine S.E., 2006, An initial map of insertion and deletion (INDEL) variation in the human genome, Genome Research, 16 (9) :1182-1190
[]Mills R.E., Walter K., Stewart C., Handsaker R.E., Chen K., Alkan C., Abyzov A., Yoon S.C., Ye K., Cheetham R.K., Chinwalla A., Conrad D.F., Fu Y., Grubert F., Hajirasouliha I., Hormozdiari F., Iakoucheva L.M., Iqbal Z., Kang S., Kidd J.M., Konkel M.K., Korn J., Khurana E., Kural D., Lam H.Y., Leng J., Li R., Li Y., Lin C.Y., Luo R., Mu X.J., Nemesh J., Peckham H.E., Rausch T., Scally A., Shi X., Stromberg M.P., Stutz A.M., Urban A.E., Walker J.A., Wu J., Zhang Y., Zhang Z.D., Batzer M.A., Ding L., Marth G.T., McVean G., Sebat J., Snyder M., Wang J., Ye K., Eichler E.E., Gerstein M.B., Hurles M.E., Lee C., McCarroll S.A., Korbel J.O., and Genomes P., 2011, Mapping copy number variation by population-scale genome sequencing, Nature, 470 (7332) :59-65
[]Pendleton M., Sebra R., Pang A.W., Ummat A., Franzen O., Rausch T., Stutz A.M., Stedman W., Anantharaman T., Hastie A., Dai H., Fritz M.H., Cao H., Cohain A., Deikus G., Durrett R.E., Blanchard S.C., Altman R., Chin C.S., Guo Y., Paxinos E.E., Korbel J.O., Darnell R.B., McCombie W.R., Kwok P.Y., Mason C.E., Schadt E.E., and Bashir A., 2015, Assembly and diploid architecture of an individual human genome via single-molecule technologies, Nature Methods, 12 (8) :780-786
[]Pugh T.J., Morozova O., Attiyeh E.F., Asgharzadeh S., Wei J.S., Auclair D., Carter S.L., Cibulskis K., Hanna M., Kiezun A., Kim J., Lawrence M.S., Lichenstein L., McKenna A., Pedamallu C.S., Ramos A.H., Shefler E., Sivachenko A., Sougnez C., Stewart C., Ally A., Birol I., Chiu R., Corbett R.D., Hirst M., Jackman S.D., Kamoh B., Khodabakshi A.H., Krzywinski M., Lo A., Moore R.A., Mungall K.L., Qian J., Tam A., Thiessen N., Zhao Y., Cole K.A., Diamond M., Diskin S.J., Mosse Y.P., Wood A.C., Ji L., Sposto R., Badgett T., London W.B., Moyer Y., Gastier-Foster J.M., Smith M.A., Guidry Auvil J.M., Gerhard D.S., Hogarty M.D., Jones S.J., Lander E.S., Gabriel S.B., Getz G., Seeger R.C., Khan J., Marra M.A., Meyerson M., and Maris J.M., 2013, The genetic landscape of high-risk neuroblastoma, Nature Genetics, 45 (3) :279-284
[]Quinlan A.R., Clark R.A., Sokolova S., Leibowitz M.L., Zhang Y., Hurles M.E., Mell J.C., and Hall I.M., 2010, Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome, Genome Research, 20 (5) :623-635
[]Rhoads A., and Au K.F., 2015, PacBio sequencing and its applications, Genomics, Proteomics and Bioinformatics, 13 (5) :278-289
[]Ross M.G., Russ C., Costello M., Hollinger A., Lennon N.J., Hegarty R., Nusbaum C., and Jaffe D.B., 2013, Characterizing and measuring bias in sequence data, Genome Biology, 14 (5) :R51
[]Seo J.S., Rhie A., Kim J., Lee S., Sohn M.H., Kim C.U., Hastie A., Cao H., Yun J.Y., Kim J., Kuk J., Park G.H., Kim J., Ryu H., Kim J., Roh M., Baek J., Hunkapiller M.W., Korlach J., Shin J.Y., and Kim C., 2016, De novo assembly and phasing of a Korean human genome, Nature, 538 (7624) :243-247
[]Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J., and Birol I., 2009, ABy SS:a parallel assembler for short read sequence data, Genome Research, 19 (6) :1117-1123
[]Sperling K., and Wiesner R., 1972, Rapid banding technique for routine use in human and comparative cytogenetics, Humangenetik, 15 (4) :349
[]Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H., Konkel M.K., Malhotra A., Stutz A.M., Shi X., Casale F.P., Chen J., Hormozdiari F., Dayama G., Chen K., Malig M., Chaisson M.J.P., Walter K., Meiers S., Kashin S., Garrison E., Auton A., Lam H.Y.K., Mu X.J., Alkan C., Antaki D., Bae T., Cerveira E., Chines P., Chong Z., Clarke L., Dal E., Ding L., Emery S., Fan X., Gujral M., Kahveci F., Kidd J.M., Kong Y., Lameijer E.W., McCarthy S., Flicek P., Gibbs R.A., Marth G., Mason C.E., Menelaou A., Muzny D.M., Nelson B.J., Noor A., Parrish N.F., Pendleton M., Quitadamo A., Raeder B., Schadt E.E., Romanovitch M., Schlattl A., Sebra R., Shabalin A.A., Untergasser A., Walker J.A., Wang M., Yu F., Zhang C., Zhang J., Zheng-Bradley X., Zhou W., Zichner T., Sebat J., Batzer M.A., McCarroll S.A., Genomes Project C., Mills R.E., Gerstein M.B., Bashir A., Stegle O., Devine S.E., Lee C., Eichler E.E., and Korbel J.O., 2015, An integrated map of structural variation in 2 504 human genomes, Nature, 526 (7571) :75-81
[]Volik S., Zhao S., Chin K., Brebner J.H., Herndon D.R., Tao Q., Kowbel D., Huang G., Lapuk A., Kuo W.L., Magrane G., De Jong P., Gray J.W., and Collins C., 2003, End-sequence profiling:sequence-based analysis of aberrant genomes, Proceedings of the National Academy of Sciences of the U-nited States of America, 100 (13) :7696-7701
[]Wang J., Yang Y., Guo S., Chen Y., Yang C., Ji H., Song X., Zhang F., Jiang Z., Ma Y., Li Y., Du A., Jin L., Reveille J.D., Zou H., and Zhou X., 2013, Association between copy number variations of HLA-DQA1 and ankylosing spondylitis in the Chinese Han population, Genes and Immunity, 14 (8) :500-503
[]Weiss L.A., Shen Y.P., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A.R., Green T., Platt O.S., Ruderfer D.M., Walsh C.A., Altshuler D., Chakravarti A., Tanzi R.E., Stefansson K., Santangelo S.L., Gusella J.F., Sklar P., Wu B., Daly M.J., and Consortium A., 2008, Association between microdeletion and microduplication at 16p11.2 and autism, New England Journal of Medicine, 358 (7) :667-675
[]Yang T.L., Chen X.D., Guo Y., Lei S.F., Wang J.T., Zhou Q., Pan F., Chen Y., Zhang Z.X., Dong S.S., Xu X.H., Yan H., Liu X., Qiu C., Zhu X.Z., Chen T., Li M., Zhang H., Zhang L., Drees B.M., Hamilton J.J., Papasian C.J., Recker R.R., Song X.P., Cheng J., and Deng H.W., 2008, Genome-wide copy-number-variation study identified a susceptibility gene, UGT2B17, for osteoporosis, American Journal of Human Genetics, 83 (6) :663-674
[]Yang Y., Chung E.K., Wu Y.L., Savelli S.L., Nagaraja H.N., Zhou B., Hebert M., Jones K.N., Shu Y.L., Kitzmiller K., Blanchong C.A., McBride K.L., Higgins G.C., Rennebohm R.M., Rice R.R., Hackshaw K.V., Roubey R.A.S., Grossman J.M., Tsao B.P., Birmingham D.J., Rovin B.H., Hebert L.A., and Yu C.Y., 2007, Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE) :Low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans, American Journal of Human Genetics, 80 (6) :1037-1054
[]Ye K., Schulz M.H., Long Q., Apweiler R., and Ning Z., 2009, Pindel:a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads, Bioinformatics, 25 (21) :2865-2871
[]Yoon S., Xuan Z., Makarov V., Ye K., and Sebat J., 2009, Sensitive and accurate detection of copy number variants using read depth of coverage, Genome Research, 19 (9) :1586-1592
[]Zerbino D.R., and Birney E., 2008, Velvet:algorithms for de novo short read assembly using de Bruijn graphs, Genome Research, 18 (5) :821-829
- 相关内容推荐





