
酚类衍生物是生物质木质素的模型化合物,也是煤焦油中的重要馏分和废水中的有害物质,通过酚类衍生物催化加氢可以制备环己酮和环己醇,产物环己酮和环己醇( KA 油) 是合成纤维尼龙 6 及尼龙 66 的单体己内酰胺和己二酸的重要原料,同时也是医药、染料等精细化学品的重要中间体[1]. 因此,酚类衍生物催化加氢制备环己酮和环己醇具有重要的科学意义和应用前景[2-3].
目前,生物圈含木质素 3×1010t 左右,每年再生 2×1010t. 木质素也是造纸工业废液中的一种主要有机成分,制浆造纸工业每年要从植物中分离出大约 5×107t 左右的木质素,但是超过 95% 的木质素仍以“黑液”直接排入河流或烧掉,只有很少一部分得到有效利用. 木质素通过化学改性,不仅在合成功能高分子领域具有潜在应用前景,而且可以从唯一的含芳烃结构的天然生物质资源出发,通过加氢制备精细化工产品如醇和酚类衍生物,及通过热裂解制取生物油,同时可以解决造纸行业因污染严重所产生的可持续发展问题[4].
木质素是一类天然芳香族高分子化合物,单体间以 C—O—C、C = C 和 C = O 键随机聚合的及其复 杂的三维网状高分子,结构片段如图1所示,没有严格的固定组成,只能粗略的分类为阔叶木木质素、针叶木木质素和草类木质素,在植物体内与纤维素、半纤维素等一起构成超分子体系. 在不同植物原料中,木质素的结构不同,即使同一原料不同部位,木质素的结构也不相同. 故木质素在结构上具有庞大性和复杂性. 木质素在化学性质上具有不稳定性,当受到化学试剂、酸度、温度变化影响时,都会发生相应的化学变化[5].【1】
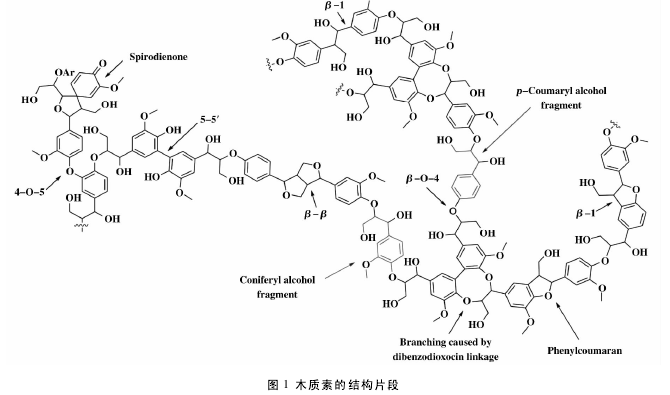
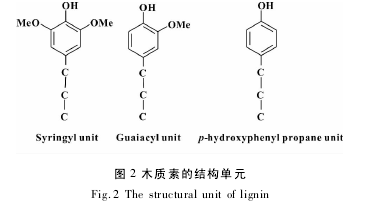
木质素结构单元间主要联接方式是 β-O-4 和 α-O-4,约占 50% 左右,其他代表性键是 β-5、β-1、5-5 !等. 木质素结构中的羟基主要是酚羟基和醇羟基,这些羟基既以游离的形式存在,也以醚键的形式和其他烷基联接. 按照植物种类不同,可分为针叶材、阔叶材和草本植物木质素 3 大类. 针叶材木质素主要由愈创木基丙烷单元构成,阔叶材木质素主要由愈创木基丙烷单元和紫丁香基丙烷的结构构成,草本植物木质素主要由愈创木基丙烷单元和紫丁香基丙烷单元及对羟基苯丙烷单元构成[6],这些单元的结构如图 2 所示.
在木质素中 O/C 和 H/C 摩尔比低于碳水化合物中的,而木质素单体碳链长度介于柴油和汽油之间,木质素可以通过热解,或催化液化得到愈创木酚等酚类化合物,酚类化合物可以通过加氢得到精细化工中间体环己醇和环己酮,也可以深度加氢脱氧制备环己烷,可极大提高木质素的利用率[7 - 8]. 经过长时间的研究,尽管木质素高值化利用已经取得了一定进展,如木质素用作混凝土减水剂、沥青乳化剂、化学固沙剂[9-10]、石油钻井液处理剂、稠油降黏剂、采油用表面活性剂、橡胶补强剂、树脂胶黏剂、土壤改良剂及农药缓释剂等[11 - 25],但仍然没有从根本上改变木质素的高值化利用现状.【2】
催化加氢是在催化剂的作用下对含有 C = C、C = O 等重键的不饱和化合物与氢加成,生成饱和化合物的反应; 氢解是在催化剂的作用下使某些单键发生裂解,形成小分子的化合物. 目前催化加氢已经广泛应用于石化行业,而且催化加氢已经发展成生物质诸如木质素及其模型物转化为化工中间体的重要方法. 木质素结构复杂,催化加氢会合成多种复杂产物,包括酚、醇、酮、烷烃等化合物,分离比较困难. 近年来,木质素加氢研究大多集中在使用木质素单体的模型化合物来研究木质素加氢反应及其机理,少数研究组则直接对木质素进行催化加氢,模型化合物的加氢分为气相状态和液相状态的多相催化加氢[26]以及均相催化加氢.
1 酚类衍生物的多相与均相催化加氢
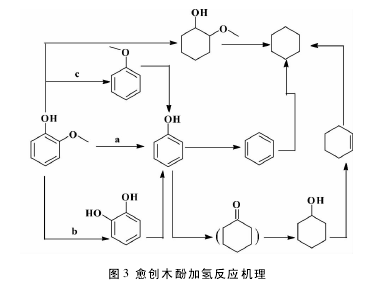
1. 1 酚类衍生物在气体状态下的多相催化加氢传统加氢是底物在气体状态下进行、高温、负载 Pd 为催化剂. Keane 等[27]以 Pd/SiO2为催化剂,150 ℃ ,对苯酚进行加氢,得到环己酮和环己醇,加入金属 Yb 形成双金属催化剂( Pd/Yb-SiO2) ,能够提高苯酚转化率和环己酮的选择性; Crisafulli等[28]对苯酚进行加氢反应研究,160 ℃,发现催化剂的活性和环己酮的选择性大小顺序 Pd/La2O3>Pd / CeO2> Pd / Al2O3,加入金属 Ca 形成双金属催化剂( Pd-Ca/Al2O3) ,明显提高了 Pd/Al2O3的催化活性和环己酮的选择性; Vishwanathan 等[29]在高温230 ℃ ,采用 Pd / MgO 和 Pd / Al2O3对苯酚加氢,发现 Pd/Al2O3加氢产物只有环己酮,Pd/MgO 产物为环己酮( 90%) 和环己醇( 10%) ,产物的选择性与载体的酸碱性有关,Al2O3酸性载体有利于环己酮的生成,MgO 等碱性载体有利于环己醇的形成;Shin 等[30]以 Pd/MgO 为催化剂,150 ℃,环己酮的选择性能够达到 95%,升高温度,苯酚的转化率和环己酮选择性都会降低,环己醇选择性会升高;L?deng 等[31]以 Mo2C / TiO2和 MoP/TiO2为催化剂,对苯酚进行加氢反应研究,350 ℃,2. 5 MPa,Mo2C / TiO2为催化剂,发生的是碳氧键的断裂,产物主要是苯,MoP/TiO2表现出对苯环强烈的加氢现象,产物为环己醇; 王铁军等[32]以愈创木酚和苯酚为原料,来尼镍( Raney Ni) 为催化剂,220 ℃,愈创木酚和苯酚转化率分别为 99% 和 90. 5%,醇的选择性都高达93%以上,该方法最大优点是不需要外界提供氢源,以 CH3OH 为氢源对模型物进行加氢,愈创木酚加氢的反应机理如图 3; 在气相状态下进行的传统多相催化加氢、高温、负载Pd为催化剂,易形成积碳,能够使催化剂失活,且产物对载体有一定的依赖性.[3]
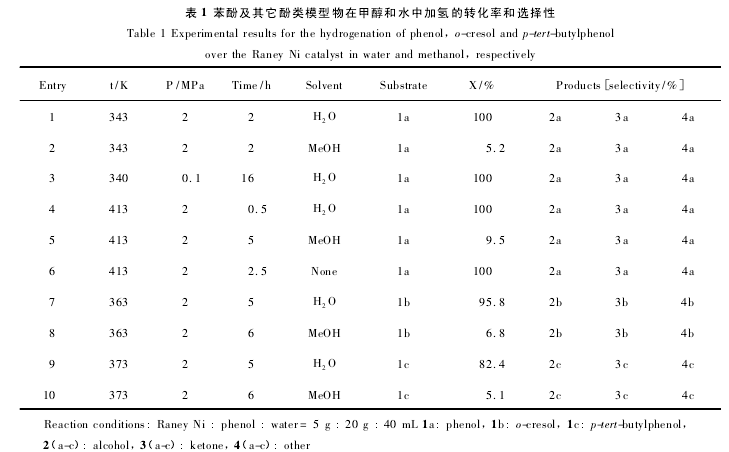
1. 2 酚类衍生物在液体状态下的多相催化加氢液相状态加氢在比较低的温度和压力下进行,节省成本与能源. Li 等[33]利用来尼镍( Raney Ni)对苯酚及其它模型物进行加氢反应研究,67 ℃,0. 1 MPa,苯酚的转化率和环己醇的选择性能够达到 99%,底物为对叔丁基苯酚时,100 ℃,2 MPa,转化率为82. 4%,该催化剂在水相中酚类的转化率和醇的选择性都优于甲醇中,该方法最大的优点是在较低温度和压力下对苯酚进行加氢,具体的转化率和选择性见表 1; 【4】
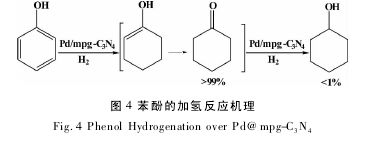
Shirai 等[34]在超临界 CO2中,55 ℃ ,H2压力 9 MPa,Rh/C 为催化剂,邻甲基苯酚的转化率达到 99%,邻甲基环己酮选择性 83%;Antonietti 等[35]以 Pd@ mpg-C3N4为催化剂,利用mpg-C3N4载体高的含氮量( 60%) 和苯酚弱酸性,对苯酚和其它木质素模型物进行了加氢反应研究,室温下 0. 1 MPa,水为溶剂,苯酚转化率和环己酮的选择性都能达到 96%以上,机理研究表明: 苯酚首先与 mpg-C3N4载体以非共平面的形式通过 O-H‥ N 结合,金属 Pd 活化氢气,接着对苯环进行逐步 加 氢, 形 成 烯 醇 式, 烯 醇 很 快 转 变 为 环己 酮,具体的反应过程如图4; 【5】
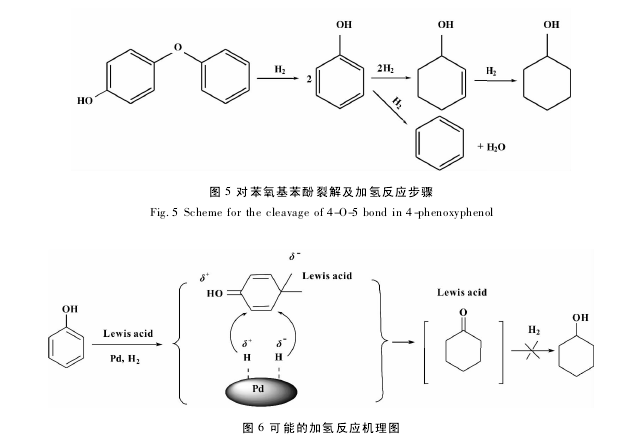
Song等[36]以Pd /XCs2. 5H0. 5PW12O40/ ACA-SO3H( X = 10,15,20,25,and 30% ) 为催化剂,以对苯氧基苯酚为模型物,发现转化率和环己醇的选择性与催化剂的酸度有关,当 X=15 时,催化剂的酸性最强,200 ℃,1 MPa 氢气压力下,对苯氧基苯酚先发生裂解生成二分子苯酚,接着对底物苯酚进行加氢,得到苯、环己烯醇,环己烯醇进一步加氢得到环己醇,环己醇的选择性能够达到 71%,具体的反应步骤如图 5; Zhao等[37]以 HCOONa/H2O 为氢源,微波辐射下对酚类模型物进行加氢反应研究,Pd/C 为催化剂,80 ℃下辐射 15 min,苯酚的转化率和环己酮的选择性达到 98%以上,与传统方法相比,具有方便,对环境无污染等优点; Han 等[38]利用 Pd/C 和 Lewis acid( AlCl3) 协同催化苯酚加氢,在30 ~50 ℃、1. 0 MPa氢气下,苯酚转化率和环己酮选择性均大于 99%,Lewis acid 具有双重功能,不仅促进苯酚加氢生成环己酮,而且可有效地抑制产物环己酮被进一步加氢生成其它副产物,反应机理如图 6; 【6】
Chen 等[39]在水中,高分子离子液体负载 Pd 为催化剂,磷钨酸为助催化剂,对苯酚及其他木质素酚类模型物进行加氢反应研究,0. 1 MPa、80 ℃,苯酚的转化率为 66%,环己酮的选择性为 98%,加入磷钨酸后,苯酚的转化率和选择性都大于99%,该方法最大优点是通过一锅法获得负载型催化剂,和对苯酚进行原位加氢; Li 等[40]以 Bu4PBr 为模板,通过化学还原法,得到了 Ce 掺杂的中空 Pd 纳米球催化剂,对苯酚进行加氢反应研究,发现 Bu4PBr 加入极大地提高苯酚的转化率,Ce 助催化剂提高了环己酮的选择性,作者还发现苯酚加氢过程中环己酮和环己醇的选择性依赖于苯酚的吸附模型,当苯酚与催化剂以共平面形式吸附生成的是环己醇,苯酚与催化剂以非共平面形式吸附有利于环己酮的生成; Li等[41]以 NiMoB/MCM-41 非晶态合金为催化剂,甲醇为溶剂,对呋喃甲醛和2-甲氧基-4-乙烯基苯酚进行了加氢反应研究,160 ℃,得到呋喃甲醇和 2-甲氧基-4-乙基苯酚,转化率分别为 72% 和 58%.Guan 等[42]利用金属有机框架( MIL-101 and MIL-53) 负载的 Pd 为催化剂,对苯酚进行了加氢反应研究,50 ℃,发现以 Pd/MIL-53 为催化剂时,苯酚转化率为 46%,环己酮选择性为 98%,Pd/MIL-101为催化剂时,苯酚的转化率为 85%,环己酮的选择性为 98. 8%.
1. 3 酚类衍生物的均相催化加氢Zhou 等[43]以[Rh( COD) Cl]2为催化剂,对苯酚和其它模型物进行了均相催化加氢反应研究,5 MPa,异丙醇为溶剂,苯酚转化率和环己醇的选择性大于98%,转化率和选择性对溶剂有一定的依赖性,当使用 THF 为溶剂时,转化率和选择性都小于25% .
我们[44]从麦草碱法制浆黑液中提取木质素,经精制后获得碱木质素; 以 NaBH4/ I2为均相催化剂,用氢气还原精制的碱木质素,首次发现 I2可以作为木质素加氢还原及氢解的均相催化剂; 并考察溶剂、温度和时间对碱木质素催化加氢效果的影响; 采用红外光谱和元素分析表征了碱木质素反应前后的结构变化; 通过自动电位滴定法测定反应前后木质素中总羟基的含量,发现反应后碱木质素中总羟基的含量为 10. 2%,较反应前增加 43. 7%;GPC 分析表明,碱木质素加氢还原后分子量分布向高分子区域扩展,数均和重均分子量增大,分子量分布明显变宽; 以配合物 Pd/PPh3和 Ni/PPh3为催化剂,二氯乙烷和乙醇的混合物为溶剂,催化加氢裂解木质素,当催化剂前体为 Ni( OAc)2和 NiCl2时,催化活性没有 Pd( OAc)2和 PdCl2的高,对产物进行粘度测试、热重分析和分子量测定,考察反应温度、时间、溶剂、催化剂前体及其用量对催化活性的影响[45].
2 结论与展望
木质素可以通过热解或催化液化得到愈创木酚等酚类化合物,酚类化合物可以通过加氢得到精细化工中间体环己醇和环己酮,可极大提高木质素的利用率. 现有的木质素模型化合物催化加氢液化工艺仍受制于催化剂的低转化率和低选择性. 传统多相催化加氢在气相状态下进行,高温高压,负载 Pd为催化剂,易形成积碳,能够使催化剂失活,且产物对载体有一定的依赖性; 目前研究最多的是液相状态下多相催化加氢,低温低压,节省能源,成本相对较低,但一些反应需要添加助催化剂,一些反应需要在特定的条件下进行. 到目前为止,对过渡金属配合物催化剂几乎是无人问津,仅有个别研究者使用( [Rh( COD) Cl]2) 为催化剂,对苯酚和其它模型物进行了加氢反应研究,因此,过渡金属与磷配体,或含其它杂原子的配体,诸如亚磷酸酯配体,膦配体,卡宾等形成的配合物为催化剂,以溶剂甲醇、甲酸钠以及 H2O 等为氢源取代氢气,及其它具有催化活性的催化剂的开发应用,对酚类化合物催化加氢制取化工中间体具有重要意义.





