
今将我们所见一家系三代色素型黄斑缺损的OCT 检查结果报告如下,以供参考。
病例摘要钱氏家系,先证者,男性,20 岁,福建平潭中楼乡人。主诉:出生后双眼视力即减退,无外伤史等,有家族遗传史(图 1)。 眼部检查:双眼视力均为 0.04,有轻度水平性眼球震颤。 散瞳验光:右眼-9.00DS/-1.00DC×40°,视力矫正 0.1;左眼-8.50DS/-1.5DC×80°,矫正无提高。 眼底:双眼视乳头色泽境界无改变,黄斑区均见一 2~4 DD 大小缺损区,病变周边清晰,中间可见灰白色巩膜及脉络膜部分血管,有广泛、不规则环形色素分布, 其余眼底及视网膜血管均显示正常。 诊断:双眼先天性色素型黄斑缺损。【图1】
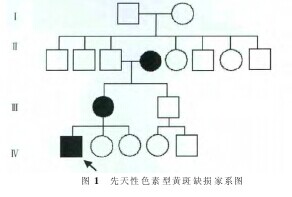
其母亲及外祖母双眼视力均在 0.02~0.04 之间,伴有轻度眼球震颤,双眼黄斑区均可见约 1~3 DD大小缺损区,并且伴有眼底鼻侧及下方广泛呈象限状的色素沉着,较对称,视力均无法矫正。
讨论先天性黄斑缺损多数属常染色体显性遗传,少数为隐性遗传,亦有散发病例。 常伴有 Down 综合症,眼部可合并其它先天性异常如虹膜缺损、视盘缺损、小角膜、小眼球等〔1〕. 眼底除了黄斑缺损以外,可伴有视网膜色素变性(RP)〔2,3〕、Leber's 先天性黑蒙,视网膜或脉络膜进行性萎缩等疾病亦见报道〔4〕. 先天性黄斑缺损真正的原因尚不清楚。 本家系先证者出生后视力即减退,无其他原因查出,家族中母亲及外祖母视力均明显减退,眼底检查证实为黄斑部缺损,呈常染色体显性遗传。国内外已有数家系先天性黄斑缺损报告。
从本家系 3 例眼底彩图,结合视力等检查诊断尚无疑问,通过 OCT 检查更显示组织学结构的明确缺损改变,从所见 3 例 OCT 成像,可明确发现先天性黄斑缺损区视网膜脉络膜组织随巩膜局限性向外凹陷,凹陷区内视网膜神经上皮层变薄,视网膜色素上皮层反射不均,证实主要为脉络膜毛细血管层的缺损。
由于 OCT 检查无创伤、易重复检查等优点,显示组织缺损定位明确,无疑为进一步丰富对该病的认识,为先天性黄斑缺损的诊断及鉴别诊断提供了一种更直观、更准确的方法。
参考文献
[1] Hayasaka Y,Hayasaka S. Bilateral congenital macular coloboma in aboy with Down syndrome[J]. Eur J Ophthalmol,2004,14 (6):565-567.
[2] 高玲,唐朝珍,姜得咏,等。原发性视网膜色素变性合并黄斑缺损兄妹二例[J]. 中华眼底病杂志,2003,19(3):187-188.
[3] Parmeggiani F,Milan E,Costagliola C,et al. Maular coloboma insiblings affected by different phenotypes of retinitis pigmentosa [J].Eye,2004,18(4):421-428.
[4] Heckenlively JR,Foxman SG,Parelhof f ES.Retinal dystrophy andmacular coloboma[J]. Doc Ophthalmol,1988,68(3-4):257-271.





