摘要:为全面揭示大头菜发酵过程中真菌群落结构及其动态变化,采用Illumina Miseq高通量测序技术对不同发酵时期工厂生产的大头菜发酵液进行真菌多样性研究。经OTU分析,24份样品中共发现4个门,19个纲,56个目,106个科,190个属,303个种,516个OTU。通过聚类分析和主成分分析发现,24个样品可根据真菌群落结构特征划分为3个聚类,将发酵的整个过程分为发酵前期、中期和后期,三个发酵阶段共有OTU数为124个。去除未分类的真菌后,所有样品中的优势菌门均为子囊菌门,相对丰度达到74.2%~99.8%,其次为担子菌门。目水平上,发酵前期炭角菌目为优势菌目,相对丰度为45.7%~60.2%,后续发酵过程中,酵母目取代炭角菌目,占据了绝对优势,相对丰度达到了63.6%~99.5%。属水平上,发酵前期明梭孢属(Monographella)为优势菌属,发酵中期和后期德巴利氏酵母属(Debaryomyces)和接合酵母属(Zygosaccharomyces)为优势菌属。
关键词:大头菜; 真菌多样性; 高通量测序;
作者简介: 吴进菊(1983-)(ORCID:0000-0002-8996-1603),女,副教授,博士,研究方向:食品加工高新技术和食品生物技术。E-mail:wujinju302@163.com;
基金: 国家自然科学基金青年科学基金项目(31501456);;湖北省高等学校优秀中青年科技创新团队计划项目(T201616) ;;湖北省普通本科高校“荆楚卓越人才”协同育人计划项目;;襄阳市科学研究开发课题(食品加工高新技术平台建设项目);;湖北文理学院食品新型工业化学科群建设项目;
Fungal Diversity of Kohlrabi during the Fermentation
WU Jinju ZENG Ruiping ZHANG Junyi ZHANG Yizhou YU Haizhong YU Bo
College of Food Science and Technology and Chemical Engineering, Hubei University of Arts and Science
Abstract:In order to reveal the diversity and community structure changes of Xiangyang kohlrabi during the fermentation, Illumina Miseq high-throughput sequencing technology was used. A total of 4 phyla, 19 classes, 56 orders, 106 families, 190 genera, 303 species and 516 OTUs were identified in the 24 samples. Both cluster analysis and principal coordinate analysis showed that the 24 samples could be divided into three clusters based on structural characteristics of fungal community, i.e. early-fermentation, middle-fermentation and later-fermentation. The total number of common OTU in the three fermentation stages was 124. After the removal of unclassified fungi, the dominant phylum in all samples was Ascomycota, with a relative abundance of 74.2% to 99.8%, followed by Basidiomycota. At the level of order, Xylariales was the dominant fungal at the early fermentation, with a relative abundance of 45.7% to 60.2%. In the subsequent fermentation process, Saccharomycetales replaced Xylariales and took an absolute advantage, with a relative abundance of 63.6% to 99.5%. At the level of genus, Monographella was the dominant fungus in the early fermentation, and Debaryomyces and Zygosaccharomyces were the dominant fungal in the middle and late fermentation.
Keyword:kohlrabi; Fungal diversity; High-throughput sequencing;
传统发酵食品制作和食用历史悠久,发酵系统开放,其中蕴藏了丰富的微生物资源[1]。近年来随着分子生物学和生物信息学的不断发展,高通量测序技术广泛地用于发酵食品微生物多样性的研究,如酸鱼[2]、意大利香肠[3]、食醋[4,5]、甜面酱[6]、发酵酒[7,8,9,10]、发酵蔬菜[11,12,13,14]、发酵豆制品[15,16,17,18,19]、发酵乳[20,21,22]等。
湖北襄阳大头菜是我国四大名腌菜之一,是襄阳市地理标志产品,具有2000多年种植史,相传是三国时期被诸葛亮发现并引入军中广泛食用,故又名诸葛菜。襄阳大头菜需经历长达数月的发酵过程才能成熟。襄阳大头菜的制作采用“三腌、五卤、六晒” 的传统工艺,在发酵过程中逐步加入食盐,大头菜成熟时发酵液中的食盐含量几乎达到饱和状态。目前,对传统发酵大头菜中微生物群落结构、种群多样性的研究相对较少,早期的研究基本是建立在传统纯培养技术研究方法上的[23,24,25],近年来有研究人员采用聚合酶链式反应—变性梯度凝胶电泳技术和高通量测序技术对襄阳大头菜腌制液膜醭进行了多样性分析[26,27,28],研究主要针对襄阳大头菜正常发酵腌制液和长膜腌制液中细菌和真菌多样性进行比较研究,找出引起发酵液长膜的主要微生物,为后续控制大头菜的品质提供一定的帮助。
本实验采用Illumina Miseq高通量测序技术对不同发酵时期的传统发酵大头菜的真菌多样性和菌群结构进行分析,从基因水平上全面揭示传统发酵大头菜发酵过程中真菌的多样性和群落结构的动态变化,对今后传统发酵大头菜的工业化、标准化生产具有极其重要的意义。
1 材料与方法
1.1 材料与试剂
大头菜发酵液样本:湖北孔明菜食品有限公司;FastPfu polymerase:北京全式金生物技术有限公司;AxyPrepDNA 凝胶回收试剂盒:AXYGEN 公司;QuantiFluor? -ST蓝色荧光定量系统:Promega公司;DL2000 DNA marker:宝生物工程(大连)有限公司;土壤基因组提取试剂盒:美国MP Biomedicals公司。
1.2 仪器与设备
9700型PCR仪:美国ABI公司;Miseq高通量测序平台:美国Illumina公司;台式冷冻离心机:德国Eppendorf公司;NanoDrop 2000 紫外可见分光光度计:赛默飞世尔科技( 中国)有限公司。
1.3 样品采集
大头菜发酵液样本由湖北孔明菜食品有限公司提供,采用“三腌、五卤、六晒”的传统工艺进行加工,按照加工工序的特点,在每一道腌制和卤制工序完成后采集样本,分别编号为1_1至1_8,每个发酵时期平行取3个样本,分别编号为a、b和c。样本采集后低温运送至实验室,置于-80℃冰箱保存备用。
1.4 样本总DNA的提取和PCR
将大头菜发酵液样本送至上海美吉生物医药科技有限公司,采用土壤基因组提取试剂盒提取总DNA。以样本中微生物总DNA为模板,以ITS1F (5′- CTTGGTCATTTAGAGGAAGTAA -3′) 和 ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′)为上下游引物扩增真菌的ITS基因序列[29]。PCR采用20μL(下同)反应体系:总DNA 10 ng,5× FastPfu buffer 4 μL, dNTP mixture (各2.5 mmol/L) 2 μL, 上下游引物(各5 μmol/L) 0.8 μL, FastPfu polymerase 0.4 μl,BSA 0.2 μL,添加双蒸水补充至20μL。PCR反应条件:95°C预变性3 min;95°C变性30 s,56°C退火30 s,72°C延伸45 s,共30个循环;72°C延伸10 min。
1.5 Illumina Miseq 测序和数据分析
将PCR产物纯化和荧光定量后,构建Miseq文库并进行双端测序,测序读长300 bp。Miseq测序得到的PE reads首先根据overlap关系进行拼接,同时对序列质量进行质控和过滤,得到优化序列。采用上海美吉生物医药科技有限公司I-Sanger云平台进行在线数据分析。采用Usearch进行OTU聚类分析,对于相似性水平大于等于97%的OTU进行生物信息统计,其中每个OTU代表一个物种[30]。采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,在各个分类水平统计样本群落的组成。利用Mothur 软件计算样本的alpha多样性,包括Chao、ACE、Shannon、Simpson和Coverage,以表征真菌群落的丰富度和多样性。采用Qiime软件计算样本beta多样性距离矩阵,使用非加权组平均法(Unweighted Pair-group Method with Arithmetic Mean,UPGMA)算法构建树状结构。
1.6 样本序列的提交及序列号
将本研究中48个fastq序列文件提交到美国国立生物技术信息中心(NCBI)SRA数据库(https://submit.ncbi.nlm.nih.gov/subs/sra),序列号为PRJNA534371。
2 结果与分析
2.1 测序结果和alpha 多样性分析
通过Illumina Miseq 高通量测序,24份大头菜发酵液样品共获得1,299,970条优化序列,平均长度从234.4bp至313.5bp(表1)。去除无重复的单序列和嵌合体后,共获得1,272,007条有效序列。经OTU分析,这些序列归属于4个门,19个纲,56个目,106个科,190个属,303个种,516个OTU。为了保证样本测序序列的均一性,对所有样品有效序列按最小样本序列数进行抽平 (每个样本30,774条有效序列)。抽平后,所有样品序列归属于4个门,18个纲,50个目,99个科,175个属,284个种,483个OTU,每个样品的OTU分类信息见表1。
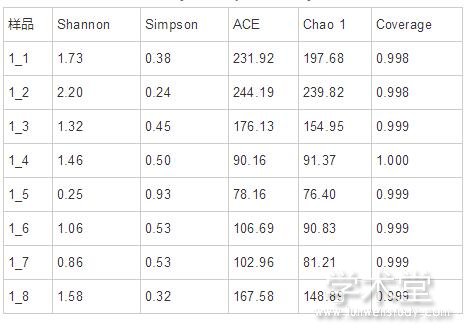
alpha多样性指数,包括Chao、ACE、Shannon、Simpson和Coverage,常用于表征在环境中微生物群落的丰度以及多样性情况。其中Chao和ACE反映群落的丰富度,Shannon、Simpson和Coverage反映群落多样性。每个样品的Coverage值均为0.998以上,表明测序深度良好,覆盖率高,可以真实展示样品中的绝大多数真菌(表2)。在样品1_1和1_2中,Chao和ACE值较高,表明样品中群落的丰富度较高。而随着发酵的进行,Chao和ACE值明显下降,而在1_8中又有所提高。分析Shannon和Simpson指数在各样品中的变化可知,群落多样性也呈现出相似的趋势。这可能是由于发酵前期加入的食盐相对较少,所以样品中真菌群落的丰富度和多样性较高,而随着发酵的进行,食盐的添加量逐渐提高,导致不耐盐的真菌逐渐消失,所以真菌群落的丰富度和多样性有所下降。而在发酵的最后时期,真菌群落的丰富度和多样性又有所提高,推测可能是有些真菌逐渐适应了高盐的环境,所以提高了真菌群落的丰富度和多样性。
表1 样品信息表 导出到EXCEL
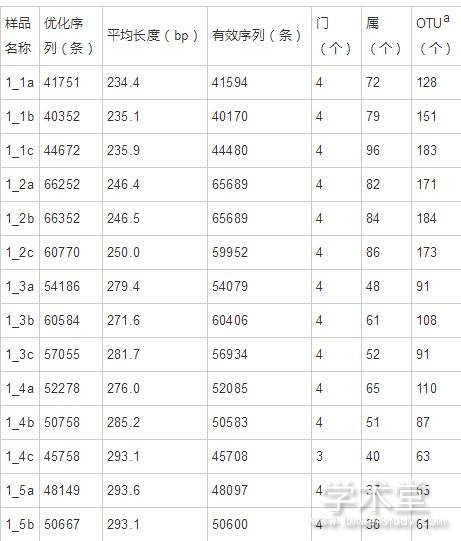
表2 alpha 多样性指数分析 导出到EXCEL
2.2 不同发酵时期大头菜发酵液样本真菌群落结构的分析
在对大头菜发酵液中不同分类学水平真菌菌群相对丰度进行分析的基础上,进一步采用样本层级聚类分析和PCA分析不同样本真菌群落结构的相似性和差异关系。在OTU分类水平上,采用bray-curtis距离算法,根据beta多样性距离矩阵进行层级聚类分析,使用UPGMA算法构建树状结构,结果如图1所示。24个大头菜发酵液样品可聚为3类,其中1_1和1_2的6个样品聚为一类,1_3、1_4、1_5的9个样品和1_6a聚为一类,1_7、1_8的6个样品和1_6b、1_6c聚为一类。由此也可将整个发酵过程大致分为三个阶段,发酵前期、中期和后期。在发酵的相同阶段,真菌群落组成结构相似。
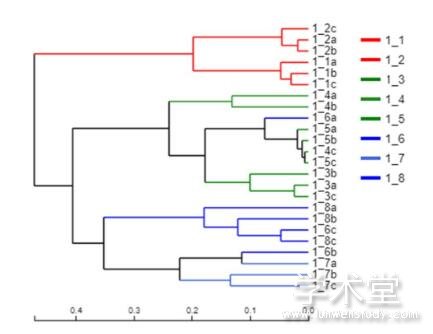
图1 OTU水平上样本真菌群落聚类分析
Fig. 1 Cluster analysis of fungal community of the samples at the OTU level
进一步采用PCA分析可知,不同样本真菌群落结构信息主要集中在前三个主成分,其累计方差贡献率为93.14%,其中PC1的贡献率为53.01%,PC2的贡献率为26.6%。以PC1和PC2作主成分分析图,结果如图2所示,样本之间的距离代表真菌群落结构的差异程度。24个大头菜发酵液样本呈现出明显的聚类趋势,除1_6a与Ⅱ类有交叉外, 1_1和1_2聚为Ⅰ类,1_3、1_4和1_5聚为Ⅱ类,1_6、1_7和1_8聚为Ⅲ类,这与UPGMA聚类分析结果一致。
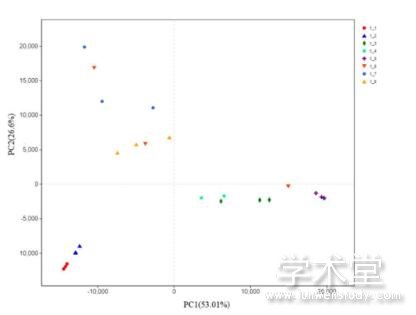
图2 OTU水平上样本真菌群落PCA分析
Fig. 2 Principal component analysis of fungal community at the OTU level
2.3 门水平真菌群落组成分析
在门水平上去除未分类的真菌后,不同发酵时期大头菜样品真菌群落组成如图3所示。去除未分类的真菌后,样品中真菌群落包含3个门,分别为子囊菌门(Ascomycota)、担子菌门(Basidiomycota)和接合菌门(Zygomycota)。其中子囊菌门为优势菌门,相对丰度为74.2%~99.8%。其次为担子菌门,相对丰度为0.25%~25.7%。担子菌门在前期和后期相对丰度较高,在中期较低。在所有样品中接合菌门相对丰度最低,在0.2%以下。
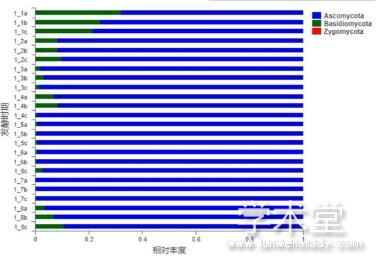
图3 门水平上真菌群落组成分析
Fig. 3 Composition analysis of fungal community at the phylum level
2.4 目水平真菌群落组成分析
不同发酵时期大头菜样品在目水平的真菌群落组成如图4所示。它们主要归属于11个菌目,包括酵母目(Saccharomycetales)、炭角菌目(Xylariales)、散囊菌目(Eurotiales)、肉座菌目(Hypocreales)、格孢菌目(Pleosporales)、煤炱目(Capnodiales)、银耳目(Tremellales )、Cystofilobasidiales目、节担菌目、norank_c__Sordariomycetes和伞型束梗孢菌目(Agaricostilbales),其它菌目相对丰度均为1%以下。
在1_1和1_2中,炭角菌目为优势菌目,相对丰度分别为60.2%和45.7%(平均值,下同),酵母目相对丰度仅为0.00%和0.07%。而在后续发酵过程中,酵母目取代炭角菌目,占据了绝对优势,相对丰度达到了63.6%~99.5%。在1_1中,银耳目相对丰度较高,为17.0%,而在1_2至1_4中降为3%以下,在1_5至1_8中降至1%以下。在整个发酵过程中,Cystofilobasidiales目在1_1中相对丰度最高,为8.4%,其次为1_4中(2.1%),在其他6个发酵时期相对丰度均为1%以下。
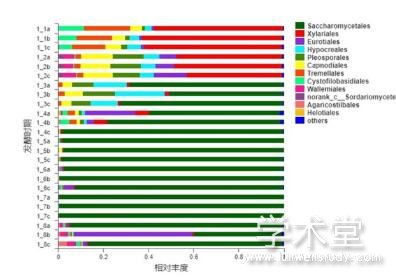
图4 目水平上真菌群落组成分析
Fig. 4 Composition analysis of fungal community at the order level
2.5 属水平真菌群落组成分析
对属水平总丰度排在前30的物种进行层级聚类,采用平均聚类方式,得到的群落Heatmap图如图5所示。Heatmap图表明了襄阳大头菜不同发酵时期样本间不同真菌菌属的相对丰度以及真菌组成的差异性和和样品间的相似性。在1_1和1_2中,明梭孢属(Monographella)为优势菌属,相对丰度分别为60.1%和45.6%。在1_3至1_5中,德巴利氏酵母属(Debaryomyces)占据了绝对的优势,相对丰度达到63.5%~96.3%。而在1_6至1_8中,接合酵母属(Zygosaccharomyces)相对丰度大大提高,发展为优势菌属,德巴利氏酵母属相对丰度有较大幅度的下降,在1_7中接合酵母属相对丰度超过德巴利氏酵母属,成为相对丰度最高的菌属(86.5%)。
由图5可知,整个发酵过程样品可聚为3类,1_1和1_2在属水平真菌群落结构比较接近,归为一类,1_3、1_4和1_5归为一类,1_6、1_7和1_8归为一类,分别对应于发酵的前期、中期和后期,这与聚类分析和PCA分析结果一致。总丰度排在前30的菌属可聚为5类,接合酵母属和德巴利氏酵母属聚为一类,在发酵前期丰度较低,而在发酵中后期丰度较高;赤霉菌属(Gibberella)、unclassified_f_Trichocomaceae、链格孢属(Alternaria)、Cystofilobasidum、Hannaella、Dloszegias、 隐球菌属(Cryptococcus)和镰刀菌属(Fusarium)聚为一类, 在发酵前期和中期相对丰度较高,而在发酵后期相对丰度较低。
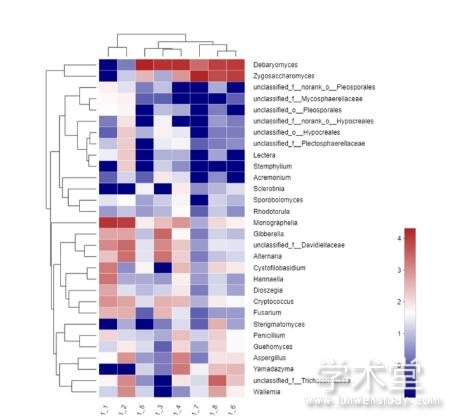
图5属水平上真菌群落组成Heatmap图。按对数值lg显示颜色,右侧为颜色梯度代表的数值。
Fig. 5 Heatmap of fungal community at the genus level. Color was displayed by the logarithm 10, and the value represented by the color gradient was on the right.
2.6 不同发酵阶段OTU Venn图分析
利用Venn图统计多个样本中共有的和独有的OTU的数量,可以直观地显示出不同分类水平样本的组成相似性和重叠程度。24个大头菜发酵液样本中共检出483个OTU,发酵前期OTU数量最多,达到329个,其次为发酵中期(266个),发酵后期样本中OTU数量最少,为246个。随着发酵的进行,OTU数量有所降低,这可能是由于在发酵过程中逐步加入了大量的食盐,抑制了不耐盐真菌的生长。从图6可以看出,三个发酵阶段共有OTU数为124个,发酵前期、中期和后期特有OTU数分别为118、58和73个,分别占各发酵阶段总OTU数的35.87%、21.80%和29.67%。
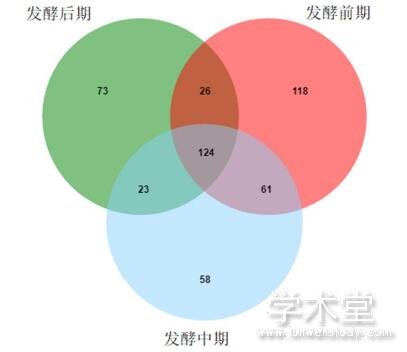
图6三个发酵阶段真菌群落OTU水平的 Veen图分析
Fig.6 Venn diagram analysis of fungal communities at the OTU level in the samples from three fermentation stages
3 结论
采用Illumina Miseq高通量测序技术对发酵过程中传统发酵大头菜的真菌多样性和菌群结构进行分析,经OTU分类学分析,24份样本中共发现4个门,19个纲,56个目,106个科,190个属,303个种,516个OTU。alpha多样性分析表明,在整个发酵过程中,前期真菌群落的丰富度和多样性更高。通过聚类分析和主成分分析发现,24个样品可根据微生物群落结构特征划分为3个聚类,将发酵的整个过程分为发酵前期、中期和后期,三个发酵阶段共有OTU数为124个。去除未分类的真菌后,子囊菌门为优势菌门,相对丰度为74.2%~99.8%。目水平上,发酵前期炭角菌目为优势菌目,相对丰度为45.7%~60.2%,后续发酵过程中,酵母目取代炭角菌目,占据了绝对优势,相对丰度达到了63.6%~99.5%。属水平上,发酵前期明梭孢属为优势菌属,相对丰度为 45.6%~60.1%。发酵中期,德巴利氏酵母属占据了绝对的优势,相对丰度达到63.5%~96.3%。而在发酵后期,接合酵母属相对丰度大大提高,发展为优势菌属,甚至超过德巴利氏酵母属。本研究全面揭示了传统发酵大头菜发酵过程中真菌群落结构及其动态变化,可为后续大头菜发酵剂的制备提供一定理论基础,对今后传统发酵大头菜的工业化、标准化生产具有极其重要的意义。
参考文献
[1]吴进菊,胡佳琪,于博,等.传统发酵食品中微生物多样性和群落结构动态变化研究进展[J].中国酿造,2016, 35(9):20-23. DOI:10.11882/j.issn.0254-5071.2016.09.005
[2] ZANG J, XU Y, XIA W, et al. Dynamics and diversity of microbial community succession during fermentation of Suan yu, a Chinese traditional fermented fish, determined by high throughput sequencing[J]. Food Research International 2018,111: 565-573. DOI:10.1016/j.foodres.2018.05.076
[3] PO?KA J, REBECCHI A, PISACANE V, et al. Bacterial diversity in typical italian salami at different ripening stages as revealed by high-throughput sequencing of 16s rRNA amplicons[J]. Food Microbiology,2015,46:342-356. DOI:10.1016/j.fm.2014.08.023
[4] GAN X, TANG H, YE D, et al. Diversity and dynamics stability of bacterial community in traditional solid-state fermentation of Qishan vinegar[J]. Ann Microbiol, 2017, 67:703-713. DOI:10.1007/s13213-017-1299-6
[5]栾春光,郝建秦,李伦,刘诗宇,王德良.高通量测序技术在食醋涨壶微生物鉴定中的应用[J].中国食品学报,2018,18(09):273-279. DOI:10.16429/j.1009-7848.2018.09.035
[6] 余丹. 基于高通量测序的传统甜面酱自然发酵过程中的微生物群落结构及其动态演替[J]. 微生物学通报, 2018, 45(5):1061-1072. DOI:10.13344/j.microbiol.china.170744
[7] SUN W, XIAO H, PENG Q, ZHANG Q, et al. Analysis of bacterial diversity of Chinese Luzhou-flavor liquor brewed in different seasons by Illumina Miseq sequencing[J]. Ann Microbio, 2016,l66:1293-1301. DOI:10.1007/s13213-016-1223-5
[8] 张苇莉, 杨慧敏, 胡景辉, 等. 朗姆酒发酵过程中微生物群落结构及其动态演替[J]. 中国酿造, 2018,37(5):60-65. DOI:10.11882/j.issn.0254-5071.20185.05.012
[9] 邢敏钰, 杜海, 徐岩, 等. 芝麻香型白酒发酵过程中乳酸菌多样性及其演替规律[J]. 微生物学通报, 2018, 45(1):19-28. DOI:10.13344/j.microbiol.china.170136
[10]郝飞, 吕锡斌, 吴耀领, 等.酱香型白酒酿造酒醅中酵母菌多样性研究[J].菌物学报, 2019, 38(5): 620-630. DOI: 10.13346/j.mycosystema.180313
[11] NGUYEN DTL, HOORDE KV, CNOCKAERT M, et al. A description of the lactic acid bacteria microbiota associated with the production of traditional fermented vegetables in Vietnam[J]. International Journal of Food Microbiology, 2013,163(1):19-27. DOI:10.1016/j.ijfoodmicro.2013.01.024
[12] LIU D, TONG C. Bacterial community diversity of traditional fermented vegetables in china[J]. LWT - Food Science and Technology, 2017,6:40-48. DOI:10.1016/j.lwt.2017.07.040
[13] LEE M, SONG JH, LEE SH, et al. Effect of seasonal production on bacterial communities in Korean industrial kimchi fermentation[J]. Food Control, 2018, 91:381-389. DOI:10.1016/j.foodcont.2018.04.023
[14] 白光剑, 邹伟, 李豪, 等.张静应用高通量测序技术分析成品甜芽菜微生物多样性[J]. 中国调味品, 2019,44(5):67-70. DOI: 10.3969/j.issn.1000-9973.219.05.016
[15] GU J, LIU T, HOU J, et al. Analysis of bacterial diversity and biogenic amines content during the fermentation processing of stinky tofu[J]. Food Research International, 2018,111:689-698. DOI:10.1016/j.foodres.2018.05.065
[16]石聪,李世瑞,李跑,等.基于高通量测序浏阳豆豉不同发酵阶段微生物多样性分析[J].食品与发酵工业,2018,44(02):27-32+39. DOI:10.13995/j.cnki.11-1802/ts.015773
[17] JI YJ, SE HL, CHE OJ. Microbial community dynamics during fermentation of doenjang-meju, traditional Korean fermented soybean [J]. International Journal of Food Microbiology, 2014, 185: 112-120. DOI:10.1016/j.ijfoodmicro.2014.06.003
[18]关统伟,向慧平,王鹏昊,等.基于高通量测序的郫县豆瓣不同发酵期细菌群落结构及其动态演替[J].食品科学,2018,39(04):106-111. DOI:10.7506/spkx1002-6630-201804016
[19]刘筱雪, 袁文娟, 丁涛, 等.基于高通量测序对四川怀远特色发酵食品微生物群落结构分析[J].四川大学学报(自然科学版),2019,56(3):537-543. DOI:10.3969/j.issn.0490-6756.2019.03.026
[20]赵顺先,关统伟,向慧平,等.基于高通量测序技术的新疆传统干奶酪乳酸菌多样性分析[J].四川农业大学学报,2018,36(05):688-695. DOI:10.16036/j.issn.1000-2650.2018.05.017
[21] BOKULICH NA, AMIRANASHVILI L, CHITCHYAN K, et al. Microbial biogeography of the transnational fermented milk matsoni[J]. Food Microbiol,2015,50:12–19. DOI:10.1016/j.fm.2015.01.018
[22]玛依乐·艾海提,西热娜依·阿布力克木,努尔古丽·热合曼.应用高通量测序法检测南疆传统酸奶中微生物多样性[J].食品科学,2018,39(20):126-131. DOI:10.7506/spkx1002-6630-201820019
[23] 杨雪, 陶兴无, 高冰, 等.发酵大头菜中乳酸菌的分离鉴定及生产初试. 中国酿造, 2008, 9:31-33
[24] 吴希茜.自然发酵腌制大头菜发酵过程中细菌的分离鉴定.山东食品发酵2011,4:11-14
[25] 吴希茜.大头菜后熟优势微生物分离鉴定及发酵剂的研制[D].重庆:西南大学,2011
[26]郭壮,沈馨,董蕴,等.襄阳大头菜腌制液膜醭细菌群落结构研究[J]. 中国酿造, 2017, 36(7):143-147. DOI:10.11882/j.issn.0254-5071.2017.07.031
[27]赵慧君,沈馨,董蕴,等.襄阳大头菜腌制液生物膜真菌多样性研究[J]. 中国调味品, 2017,42(12):61-65. DOI:10.3969/j.issn.1000-9973.2017.12.013
[28] 赵慧君, 葛东颖, 刘倩, 等. 襄阳大头菜不同发酵状态腌制液中细菌多样性分析[J]. 中国调味品, 2018,43(9):44-48. DOI:10.3969/j.issn.1000-9973.2018.09.009
[29] GARDES M, BRUNS TD. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts[J]. Molecular Ecology, 1993, 2(2): 113-118. DOI:10.1111/j.1365-294X.1993.tb00005.x
[30] EDGAR RC. Highly accurate OTU sequences from microbial amplicon reads[J]. Nat Method, 2013, 10(10): 996-998. DOI:10.1038/NMETH.2604





