两种HLA分型方法对比
摘要:对比两种HLA分型方法的准确率, 通过使用polysolver方法和研究出的新方法这两种方法对100个个体的全基因组外显子测序数据 (WES) 进行分型, 然后将两种方法的分型结果与这100个个体的sanger一代测序结果进行比较, 找到新方法的优点和不足, 并且为动物MHC基因研究提供算法基础。经过新方法的开发以及优化, 最终与polysolver方法的结果进行比较, 得出结论, 发现研究的新方法与polysolver分型方法相比准确率相对较高, 且运行时间相对较短。两种方法均只能对HLA一类分子进行分型, 这一点在将来实验中会进一步改进。
关键词:HLA分型; polysolver; 分型新方法; 高通量测序数据;
作者简介: 原应博 (1993-) , 男, 黑龙江八一农垦大学生命科学技术学院2017级硕士研究生。; *方铭 (1979-) , 男, 教授, 博士研究生导师, E-mail:fangming618@126.com;
收稿日期:2019-03-22
基金: 国家自然科学基金面上项目 (31672399和31872560);
Comparison of Two HLA Typing Methods
Yuan Yingbo Fang Ming
Heilongjiang Bayi Agricultural University JiMei University
Abstract:Comparing the accuracy of two HLA typing methods, we typed the whole genome exon sequencing data (WES) of 100 individuals by using the polysolver method and the new method developed. Then we compared the results of the two methods with those of the Sanger generation of these 100 individuals, and found that the new method is somewhat inadequate. Moreover, it also provides an algorithmic basis for animal MHC gene research. After the development and optimization of the new method, the results of the new method are compared with those of the polysolver method. We found that the accuracy of the new method is relatively higher than that of the polysolver typing method, and the running time is relatively shorter. Both methods can only classify HLA Ⅰ-class, which will be further improved in the future.
Keyword:HLA typing; polysolver; the new typing method; high throughput sequencing data;
Received: 2019-03-22
MHC区域是人类基因组中基因最密集的区域之一, 位于人类6号染色体的短臂上[1]。而人类白细胞抗原 (HLA) 基因是MHC区域中研究最多的基因, 它编码负责抗原肽在适应性免疫应答中呈递的细胞表面蛋白[2]。
人类白细胞抗原 (HLA) 基因与许多自身免疫和感染性疾病表型相关, 是区别自身和非自身之间免疫学中的重要因素, 从而形成相关的数据库[3,4,5]。由于HLA基因在免疫学中的关键作用, HLA分型在临床背景下被广泛用于器官和造血干细胞移植中的供体和受体的匹配[6,7]。因此, 确定HLA基因的等位基因状态 (HLA分型) 对于群体测序项目中进行样品的免疫遗传表征检测是非常重要的。然而, HLA基因具有超多态性和序列相似性, 这使得准确的推断HLA基因成为一个具有挑战性的问题。
目前用于HLA等位基因的高分辨率分型的金标准, 基于序列的分型 (SBT) , 使用Sanger测序或HLA基因的靶向扩增, 然后进行下一代测序。在现中使用的金标准为sanger测序的结果[2,8]。
现对两种HLA分型方法进行简单的比对, 分别是polysolver和另一种新开发的算法。两种方法均对100个个体的全基因组外显子测序数据 (WES) 进行HLA分型, 通过与金标准对比来比对他们的结果。Sachet A Shukla等[9]的polysolver方法与新方法首先都是将WES数据与国际免疫遗传学数据库 (IMGT) 中的已知HLA基因文库对齐提高读数的精确性[10], 之后使用novoalign软件进行对齐, polysolver方法后续使用贝叶斯方法进行分型, 而新方法对对齐之后的数据建立比对矩阵, 通过一对等位基因在测序时匹配的read片段个数综合之后进行迭代比对的方法进行分型。通过两种不同的方法对实验个体的测序数据进行分型, 对分型结果进行比对, 验证新方法的实验结果, 同时比较两种方法的优劣, 为动物MHC基因研究提供算法基础。
1 两种方法介绍
1.1 背景介绍及方法来源
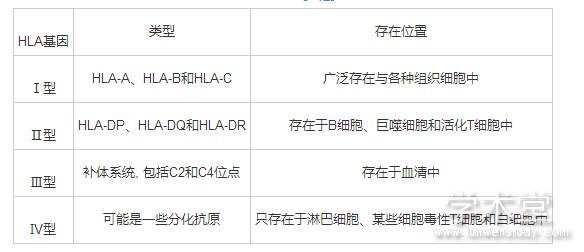
主要组织相容性复合体 ( major histocompatibility complex, MHC) 是一组编码动物主要组织相容性抗原的基因群的统称[11]。人类的MHC位于人的6号染色体短臂上, 小鼠的MHC位于小鼠的17号染色体上。MHC的长度大约为4×106bp。人类的MHC也叫做HLA (human leukocyte antigen, HLA) 复合体。小鼠的MHC则被称为H-2基因。由于MHC的多基因特性, 依据其编码分子的结构、组织分布与功能差异, 可分为MHC I类、MHC II类、MHC III类基因, 分别编码MHC I类分子、MHC II类分子、MHC III类分子[12]。人类的MHC通常被称为人类白细胞抗原 (human lymphocyte antigen, HLA, 以下简称HLA) , 位于第6染色体短臂6p21.31 区域, 占人类基因组的1/30[13,14]。HLA基因主要分为四种类型, 如表1。
通过改进HLA分型技术, 不仅有利于快速准确地找到合适供者, 大大地提高骨髓库的使用率, 使其更好的服务于患者, 而且可以为HLA的科学研究与技术创新提供基础性的数据支持, 并且通过一系列的改进和完善, 也可以为动物MHC基因研究提供算法基础。
表1 HLA基因类型 导出到EXCEL
随着测序技术的不断发展, HLA分型技术, 也逐渐从传统的血清学分型和细胞学分型转变为DNA分型[15]。传统的血清学分型主要借助的是微量淋巴细胞毒试验或称为补体依赖的细胞毒试验[16]。细胞学分型主要是通过纯合分型细胞 (homozygote typing cell, HTC) 及预致敏淋巴细胞试验 (PLT) 对HLA分型[17,18,19]。DNA分型方法主要分为两种:基于核酸序列识别的方法和基于序列分子构型的方法[20]。
但是, 由于传统的血清学方法、细胞学方法和DNA分型方法均需要动手实验, 操作一系列的实验器材, 调整所需要的一系列实验试剂, 这一过程相较于采用软件分型来说比较繁琐和浪费时间, 并且如果在实验过程中操作失误, 或者实验试剂配制出现误差, 对结果的影响是非常大的, 增加了实验的工作量, 浪费了宝贵的时间。所以, 如果采用计算机软件分型方法将会大大减少人们的工作量, 并且节省大量时间和实验所需材料。基于此, 笔者研发了一种新型的分型方法, 致力于减少HLA分型的工作量以及工作时间。同时, 将这种方法与Sachet A Shukla等[9]的polysolver方法进行对比。Polysolver的方法主要采用贝叶斯分类方法对HLA进行分型。这两种方法均可以对HLA一类基因进行分型。
在进行结果比对时, 需要一个金标准作为衡量结果的指标, 在HLA分型中, PCR-SBT测序方法是现世界卫生组织 (WHO) 推荐的HLA分型方法的“金标准”[21]。而PCR-SBT测序方法就是平常所说的一带测序方法。笔者采用Sanger法进行测序, 获得这100个个体的分型结果作为金标准。
两种方法的输入数据均为全基因组外显子测序数据, 对于实验的操作以及前期数据的收集均有帮助, 并且对于实验后期结果的比较也减少了误差, 因此, 选择这两种方法进行对比。
1.2 方法介绍
1.2.1 数据来源
本次实验数据主要由华大基因提供, 在其提供的众多全基因组外显子测序数据中随机抽取100个个体作为本次实验的主体。同时, 由华大基因提供这100个测序数据的sanger测序结果作为金标准。同时, 由华大基因提供服务器来支持本次实验的顺利进行及结果比较。
数据库数据由国际免疫遗传学数据库 (IMGT) 提供多序列比对文件 (MSA) , 通过对多序列比对文件进行筛选合并等操作, 以此来构建已知HLA等位基因的全长基因组参考文库[22]。IMGT, 国际免疫遗传学数据库, 是一个专注于所有脊椎动物物种的免疫球蛋白、T细胞受体和主要组织相容性复合体的整合数据库, 由Marie-Paule Lefranc、法国科学研究中心、法国蒙彼利埃第二大学发起并共同协调[23]。 IMGT包括两个数据库:LIGM-DB (面向免疫球蛋白和TcR) 和MHC/HLA-DB。IMGT由专家注释的序列和比对表组成[22]。LIGM-DB包含了来自78种物种的超过19000个免疫球蛋白和TcR序列。MHC/HLA-DB包含了I类和II类HLA比对表。一个为免疫球蛋白、TcR和MHC序列比对而开发的IMGT工具DNAPLOT也是可用的。IMGT与EMBL数据库紧密合作。IMGT的目标是建立一个对所有免疫遗传学数据的通用访问, 包括序列、寡核苷酸引物、基因图谱和免疫球蛋白、TcR和MHC分子的其他遗传数据, 并提供一个图形化的用户友好的数据访问[10,22]。IMGT将对医学研究 (自身免疫病、艾滋病、白血病、淋巴瘤) 、治疗方法 (抗体工程学) 、基因组多样性和基因组进化研究具有重要影响。因此, 选择国际免疫遗传学数据库。
1.2.2 Polysolver方法介绍
Polysolver方法首先将WES数据与MHC区域的严格过滤序列进行比对, 筛选出其中的HLA基因测序数据, 然后通过novoalign比对软件将筛选出来的HLA测序数据与IMGT数据库中的HLA基因进行对齐, 将测序数据比对在HLA基因上, 最后通过贝叶斯算法, 根据比对在每个HLA基因上的测序数据、插入序列的大小以及每个种族的各个等位基因的先验概率来计算该基因的似然值, 通过比较, 得出第一个等位基因类型, 然后根据第一个等位基因来得出第二个等位基因类型, 从而得到该个体的HLA基因分型结果。如图1所示。
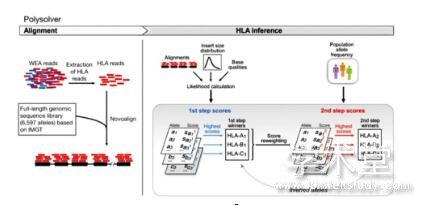
图1 polysolver流程图
1.2.3 新方法介绍
笔者为新方法构建了一个数据库。为了更大限度的检索所有的HLA读数, 笔者基于IMGT数据库中的多序列比对文件 (MSA) 构建了已知HLA等位基因的全长基因组参考文库[24]。
新方法的主要步骤包括两步, 首先, 对在WES数据中对HLA数据进行比较准确的提取。在这一步中, 笔者先通过序列比对软件将WES数据中的MHC区域提取出来, 之后将提取出来的区域与之前构建好的IMGT参考文库进行比对来提高比对的精度。笔者在第一步中使用samtools和Novoalign两个软件进行比对, 设定好参数使每个HLA读数都是最佳的[25,26]。
然后, 根据提取HLA数据对其进行分型。在这一步中, 首先对上一步提取的HLA数据进行统计, 统计每一个HLA所比对上的reads个数以及read种类, 其次根据统计结果构建一个HLA基因与测序片段 (reads) 的关系矩阵, 其中列名为reads名称, 行名为HLA基因名称, 对应的矩阵位点如果是0, 则表明该read未比对到对应的HLA上, 如果是1则表明该read比对到相应的HLA, 如表2所示。之后通过迭代对比匹配的方法进行分型。
表2 HLA与测序片段关系矩阵 (例) 导出到EXCEL

迭代匹配的方法如下所示:
(1) 根据关系矩阵, 尽心简单的统计获得比对在每一个HLA基因的read个数, 通过排序比较, 获得一个比对read个数最多的HLA基因, 从而得到一对等位基因的第一个基因, 假设为A。以表1为例, 三个hla中hla01号有3个read比对上, hla02号有4个, hla03号有2个, 那么此时A则为hla02。
(2) 笔者根据A, 通过关系矩阵和穷举法, 使A与每一个HLA基因进行比较, 即关系矩阵的每一行进行比较, 比较方法为将A所在的行与选定的行进行统计, 统计出两行均为1的列的个数, 即均比对到两个HLA上read的个数, 假定为X, 以及选定行独有1的列数, 即仅比对到选定行HLA的read的个数, 假定为B1, 通过求B1与1/2的X的和来计算在有A为先验影响的条件下, 选定行HLA的比对的read个数, 假定为sumread。比较每一次统计的sumread结果, 筛选出一对等位基因, 从而得到另一个基因, 假设为B。此时初步分型结果为A, B。
(3) 根据步骤 (2) 得到另一个基因B, 采用步骤 (2) 的方法, 得到选定的基因C。如果基因C与A不同, 重复步骤 (2) , 得到基因D, 此时分型结果为B, D, 进入步骤 (4) 。如果此时基因C与基因A相同, 那么分型结果为B, C, 即B, A。
(4) 根据步骤 (3) 的分型结果得到的另一个基因为D, 迭代重复步骤 (3) , 得到最后的分型结果。
步骤 (2) 的公式如下图所示,

图2 算法公式
新方法流程图如下图所示:
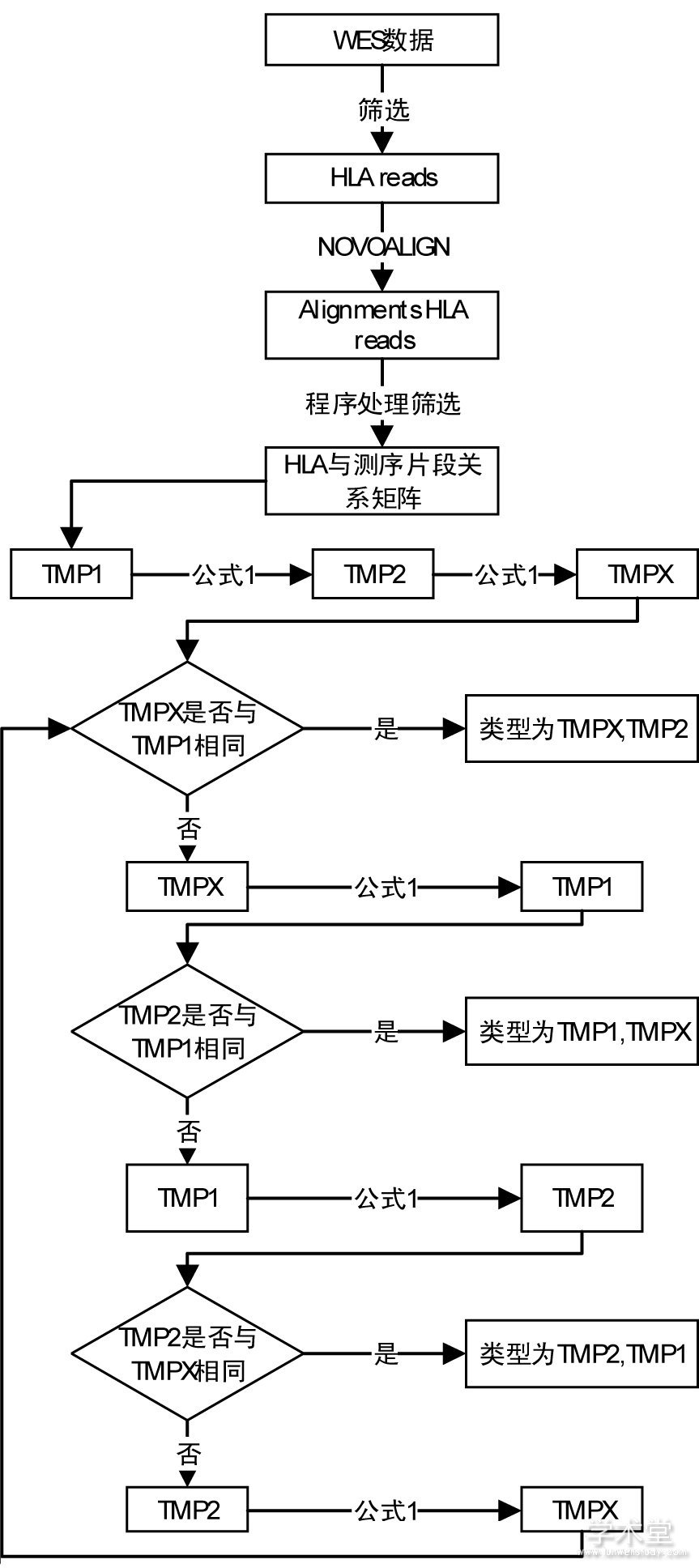
图3 新方法流程图
1.3 相关软件环境介绍
两种方法据需要在linux环境下运行, 要用到samtools软件, novoalign软件, GATK软件以及perl软件, 其中Perl需要bioperl、math::basecacl、POSIX等一些模块来运行。采用samtools软件对全基因组测序数据进行格式转换以及初步的数据比对;使用novoalign软件对samtools软件的处理结果进行进一步的数据比对和筛选, 使之更加准确;GATK全称是The Genome Analysis Toolkit, 是Broad Institute开发的用于二代重测序数据分析的一款软件, 里面包含了很多有用的工具, 主要注重与变异的查找, 基因分型且对于数据质量保证高度重视[21]。软件的主体是由perl语言写的, 需要用到其中关于数据处理以及生物信息格式文件的处理模块。通过以上的组合, 实现软件的运行。
1.4 两种方法的简单使用
首先在LINUX环境中将所需要的依赖环境配置好, 同时将所需要的软件安装到服务器上。通过软件提供的测试用全基因组外显子测序数据进行简单测试和调试, 是软件可以正常运行。配置好运行环境以及测试文件可以运行之后, 就可以进行试验测试了。
两种方法使用的时候均需要输入WES测序数据。通过写脚本使实验数据可以批量运行, 由于使用的华大基因的服务器, 可以将测试任务递交到后台自动运行, 要注意服务器的承载限度, 递交任务之后继续优化程序同时静待分型结果, polysolver分型结果输出在winnerhla.txt文件中, 新方法的分型结果输出在besthla.txt中。
2 两种方法比较
通过对100个个体的进行分型, 分型结果统计如表3。
表3 分型结果统计表 导出到EXCEL
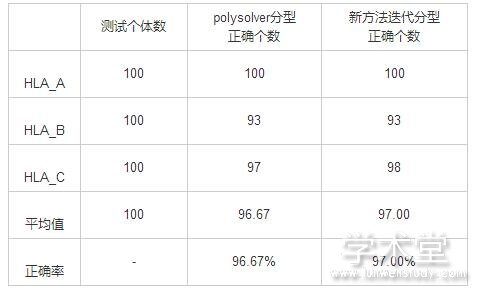
从表3笔者可以发现在对HLA的A类分子进行分型的时候, 两种方法的分型结果均正确。在对HLA的B类分子进行分型的时候, 两种方法均出现错误, 笔者初步判断错误是由于纯合子或者测序数据与数据库版本不匹配造成的, 后续通过进一步实验来查明原因。在对HLA的C类分子进行分型的时候, polysolver方法正确97个, 而新开发的方法正确为98个, 有了改进。在HLA分型中, 由于HLA分子种类繁多, 其分型的难度也是一定的, 仅仅改进一个个体也是很大的改进。假如基础实验个体不仅仅有100个, 而是上万个甚至百万个的时候, 这个改进可以大大降低分型错误率。
汇总以上三种分子的分型结果, 笔者可以发现, 采用新方法迭代分型的话正确率平均值为97%高于使用polysolver分型的96.67%。
在时间方面, 由于软件的运行时间会受服务器状态的影响, 实验采用的服务器由华大基因提供天津超算的接口, 而服务器不仅仅只有一个人在用, 并且服务器运行程序越多运行越慢, 对时间的影响比较严重。因此, 考虑到服务器的运行状态及其承载限度, 对时间并未做详细的统计。但是在运行过程中, 笔者粗略的比对了一下运行时间, 根据表现来比较, 新方法相较于polysolver方法要快一点。
3 结论与讨论
通过比较, 新方法在准确率上比polysolver方法要好一点, 在为HLA-C分型时, 新方法相较于polysolver方法有了改进。在运行时间方面, 根据两个方法的表现, 新方法要略快与polysolver方法, 运行时间会随服务器型号以及服务器的运行状态来决定。因此, 在运行时间方面的比较未作详细统计。
通过新方法改进分型, 使得HLA分型的准确率得到了提升, 尽管只有零点几的百分点, 但是对于HLA分型来说是一个很大的改进, 同时为HLA分型的软件方法又添加一项技术支持。但是, Polysolver方法与新方法都仅仅只能对HLA Ⅰ- class进行分型, 同时输入的数据要求为全基因组外显子测序数据。针对该项不足, 笔者对新方法正在进行改进, 未来可以对HLA基因的Ⅱ- class也可以分型。而且polysolver方法的数据库无法更新。新方法的数据库需要手动更新, 针对手动更新这一点正在改进, 将来可以自动更新。
HLA分型仍然是现在医学的难题, 特别是在现在短读长测序依旧盛行的时代。虽然三代测序可以有效解决HLA的测序, 但现阶段成本较低的依旧是以illumina为首的测序分型。当前市面的一些HLA分型开源软件基本可以分为两种, 一种是基于WGS、WES、target-seq的软件, 包括xhla、SOAP-HLA、optitype等。另一种是基于SNP芯片的软件, 包括SNP2HLA、eHLA等, 基于snp的分型准确率需要参考人群专一, 比如中国北方汉族和中国南方汉族都不能用一个参考数据训练。而且分型的准确率较差, 不能具有实用价值, 一般只能分到四位。
相对于其他分型软件, 新方法主要采用迭代比较的方法, 不断的改进每次比较的先验概率, 并且反过来验证前一次推断的结果, 从而使得分型的结果更加准确。并且, 可以根据IMGT官网最新数据来更新HLA等位基因数据库。同时可以为动物MHC基因研究提供算法基础, 通过更改数据库等操作实现为动物等其他物种分型。
参考文献
[1] Cao Hongzhi, Wu Jinghua, Wang Yu, et al.An integrated tool to study MHC region:accurate SNV detection and HLA genes typing in human MHC region using targeted high-throughput sequencing[J].PloS one, 2013, 8 (7) :e69388.
[2] Dilthey Alexander T, Gourraud Pierre-Antoine, Mentzer Alexander J, et al.High-Accuracy HLA Type Inference from Whole-Genome Sequencing Data Using Population Reference Graphs[J].PLOS Computational Biology, 2016, 12 (10) :e1005151.
[3] International Multiple Sclerosis Genetics Consortium, Beecham Ashley H, Patsopoulos Nikolaos A, et al.Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis[J].Nature Genetics, 2013, 45:1353-1360.
[4] The Australo-Anglo-American Spondyloarthritis Consortium, the Wellcome Trust Case Control Consortium, Evans David M, et al.Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility[J].Nature Genetics, 2011, 43:761-767.
[5] Genetic Analysis of Psoriasis Consortium, the Wellcome Trust Case Control Consortium, Strange Amy, et al.A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1[J].Nature Genetics, 2010, 42:985-990.
[6] Morishima Yasuo, Sasazuki Takehiko, Inoko Hidetoshi, et al.The clinical significance of human leukocyte antigen (HLA) allele compatibility in patients receiving a marrow transplant from serologically HLA-A, HLA-B, and HLA-DR matched unrelated donors[J].Blood, 2002, 99 (11) :4200-4206.
[7] Lee Stephanie J, Klein John, Haagenson Michael, et al.High-resolution donor-recipient HLA matching contributes to the success of unrelated donor marrow transplantation[J].Blood, 2007, 110 (13) :4576-4583.
[8] 张瑞丽, 杜国栋, 杨玉梅, 等.一种新的HLA分型方法的介绍[J].生物医学工程与临床.2017 (1) :103-105.
[9] Shukla Sachet A, Rooney Michael S, Rajasagi Mohini, et al.Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes[J].Nature Biotechnology, 2015, 33 (11) :1152-1158.
[10] Robinson James, Halliwell Jason A, McWilliam Hamish, et al.The IMGT/HLA database[J].Nucleic acids research, 2013, 41 (Database issue) :D1222-D1227.
[11] Rammensee H, Bachmann J, Emmerich N P, et al.SYFPEITHI:database for MHC ligands and peptide motifs[J].Immunogenetics, 1999, 50 (3-4) :213-219.
[12] Pamer Eric, Cresswell Peter.Mechanisms of MHC class I-restricted antigen processing[J].Annual Review of Immunology, 1998, 16 (1) :323-358.
[13] Althaf Mohammed Mahdi, Kossi Mohsen El, Jin Jon Kim, et al.Human leukocyte antigen typing and crossmatch:A comprehensive review[J].World Journal of Transplantation, 2017, 7 (6) :339-348.
[14] Horton R, Wilming L, Rand V, et al.Gene map of the extended human MHC[J].Nature Reviews Genetics, 2004, 5 (12) :889-899.
[15] 刘川.HLA基因分型方法的进展[J].实验与检验医学, 2011, 29 (3) :261-262.
[16] Chapuis A G, Roberts I M, Thompson J A, et al.T-Cell Therapy Using Interleukin-21-Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression[J].Journal of Clinical Oncology Official Journal of the American Society of Clinical Oncology, 2016, 34 (31) .
[17] 付东杰.HLA的分子生物学检测的临床分析[J].医药前沿, 2014 (10) :245-246.
[18] 邹森, 洪坤学.基于二代测序技术的HLA基因分型进展[J].检验医学与临床.2017, 14 (1) :144-146.
[19] 匡国杰, 许晓光, 肖漓, 等.肾移植受者抗HLA抗体的研究进展[C].全国免疫学学术大会, 2015.
[20] Alcántara Montero A, Sánchez Carnerero C I, Ibor Vidal P J, et al.CDC guidelines for prescribing opioids for chronic pain[J].Semergen.2017, 43 (4) :e53.
[21] 黄飞, 肖露露, 阎文瑛, 等.PCR-SSP/PCR-SBT-HLA高分辨等位基因分型比较[J].中山大学学报 (医学科学版) .2006, 23 (s1) :21-24.
[22] James Robinson, Waller Matthew J, Peter Parham, et al.IMGT/HLA and IMGT/MHC:sequence databases for the study of the major histocompatibility complex.[J].Nucleic Acids Research.2003, 31 (1) :311-314.
[23] Giudicelli V, Chaume D, Lefranc G, et al.IMGT[C].The international ImMunoGeneTics database, 2003.
[24] James Robinson, Kavita Mistry, Hamish Mc William, et al.The IMGT/HLA database[J].Nucleic Acids Research.2013, 409 (1) :43.
[25] Li Heng, Handsaker Bob, Wysoker Alec, et al.The Sequence Alignment/Map (SAM) Format and SAMtools[J].Bioinformatics, 2009, 25 (1 Pt 2) :1653-1654.
[26] Schbath Sophie, Martin Véronique, Zytnicki Matthias, et al.Mapping Reads on a Genomic Sequence:An Algorithmic Overview and a Practical Comparative Analysis[J].Journal of Computational Biology A Journal of Computational Molecular Cell Biology, 2012, 19 (6) :796-813.





