近年来,手性制药工业迅速发展壮大,单一对应体药物每年以大于 20% 的速度增长,其对手性胺的需求也随之增长。目前,超过 70% 的药物都是手性胺及其衍生物,如神经类药物、心血管药物、抗高血压药物、抗感染药物及疫苗等的合成都是以手性胺作为中间体,抗糖尿病新药 Janu-via 的主要成分西他列汀是 R-型胺。这就催促人们寻找一种高效制备手性胺的方法,而转氨酶的出现使研究者看到了曙光。转氨酶以其高选择性、高转化率及温和的反应条件赢得了广大研究者的青睐,其能够催化 1 个氨基供体( 氨基酸或简单的胺) 上的氨基转移到前手性的受体酮,得到手性胺和副产物酮或者 α - 酮酸( 图 1) ,反应过程需要磷酸吡哆醛 ( pyridoxal phosphate,PLP) 的参与。
转氨酶合成手性化合物已经发展为一项关键的不对称合成技术,受到越来越受到众多研究者的重视和关注。2013 年 2 月 28 日,第一届国际转氨酶生物催化研讨会于瑞典斯德哥尔摩召开,鉴于此,本文作者将转氨酶在手性化合物合成中涉及到的蛋白工程、表达与固定化、进程工程及应用方面的研究现状予以综述。
1 转氨酶蛋白工程
蛋白工程是指通过改变已知蛋白的结构来改变其性质的过程,需要借助计算机和生物信息学手段来实现,是研究酶的功能及进化的重要技术,也是改变酶性质、开发新酶的重要方法。利用蛋白工程的方法对转氨酶进行改造即为转氨酶蛋白工程,其目的是得到有工业应用价值的酶,打通手性药物及化工产品的酶法合成途径。所用关键技术包括理性设计、定向进化以及两者组合方法; 所涉及工具主要包括迭代饱和诱变( it-erative saturation mutagenesis,ISM ) 、组合活性位点饱和测试( combination of active site saturationtest,CASTing ) 、蛋白序列活性关系 ( protein se-quence activity relationship,ProSAR) 等。随着计算机技术以及生物信息学的快速发展,转氨酶蛋白工程迎来了第三次浪潮,成为国内外研究热点。
转氨酶蛋白工程主要包括 5 个步骤: 选择合适的模板、稳定模板、确定活性位点、活性位点周围氨基酸残基理性设计或定向进化、突变体活性评估。经过以上几步,就可以根据人们的意愿将一个原本对某一底物无活性的转氨酶变为对此底物活性很高的生物催化剂,在此方面最成功的例子当属 Savile 等利用蛋白工程手段得到的突变型 ATA-117,其能够用于工业化生产西他列汀。到目前为止,此酶是转氨酶工业化应用最成功的例子,利用该酶的西他列汀生物催化合成工艺获得 2010 年美国总统绿色化学挑战奖。目前,国内外很多课题组都在进行转氨酶蛋白工程的研究,近五年来,新型转氨酶如雨后春笋般涌出( 表 1) 。
2 转氨酶表达与固定化
转氨酶的表达目前有 3 种途径。第一,利用原始菌株表达,由于原始菌株表达量低及效率低下等不足,此途径现一般只在新产酶菌株初步筛选阶段使用; 第二,利用工程菌表达,此法应用最多,目前转氨酶已经在原核与真核系统中实现表达。原核表达普遍采用大肠杆菌表达系统,例如 Iwasaki 等成功构建一种 R-型转氨酶( R-TA) 的表达工程菌 E. coli HB101-pAT28-R-TA,通过工程菌的诱导表达可以得到大量 R-TA; Savile等构建工程菌 E. coli W3110-pCK110700-ATA-117 表达 ATA-117 及突变酶; Cassimjee 等利用工程菌E. coli BL21 ( DE)-pET28-ATA-113 成功实现转氨酶 ATA-113 的大量表达。原核表达系统是目前掌握最为成熟的表达系统,其优点在于能够在较短时间内获得基因表达产物,而所需的成本相对低廉。但原核表达系统还存在许多难以克服的缺点,如无法对表达时间及表达水平进行调控,目的蛋白易形成包涵体,导致产物纯化困难。为弥补以上不足,许多学者将原核基因调控系统引入真核基因调控领域,采用真核系统进行目标蛋白的表达。真核表达有酵母表达系统、昆虫细胞表达系统和哺乳动物细胞三种系统,目前,转氨酶多采用毕赤酵母表达系统,如 Bea 等成功构建了ω-TA 的真核表达系统 P. pastoris GS115-pPIC-ω-TA; Weinhandl 等通过转氨酶基因密码子优化,构建了能够大量表达 ilvE 的 P. pastoris Mut-SpPpT4_S / pPpT4 _GAP_S-ilvE。真核表达系统能诱导基因高效表达,可达原始表达量的 105 倍,另外,其还能严格调控基因表达,是原核表达系统所不能及的,因此,利用真核表达系统来表达目的蛋白越来越受到重视,其多用于可调控的转氨酶的大量表达。第三,通过无细胞体系表达。Kwon等建立了一种无细胞蛋白合成系统成功实现了 ω-TA 的非克隆性表达,此方法主要使用于转氨酶的高通量筛选。
在转化中作者通常用到 2 种酶形式: 游离酶和固定化酶。游离酶活性虽高但是只能用 1 次,且会影响产物的分离纯化,而固定化酶则会大大提高重复利用率,因此成为了研究重点。转氨酶的固定化包括固定化细胞和固定化游离酶。固定化细胞所用材料主要是壳聚糖、海藻酸钠以及一种新型材料 LentiKats。Rehn 等发现利用壳聚糖固定化含有 ω-TA 的大肠杆菌细胞,在优化条件下,细胞加载量可达 3. 2 g·g- 1壳聚糖,残留活性大于 60%,在 1 h 的反应中,连续 8 次反应,固定化酶活性仍大于 90%。Fernandez 等利用LentiKats 固定化含有 ω-TA 的大肠杆菌细胞,其稳定性得到了提高,在连续五次合成 1-苯乙胺及3-氨基-丁苯后,残留活性仍大于 80% 。固定化游离酶所用材料主要有聚合树脂、溶胶/硅藻土基质、壳聚糖、硅胶。Truppo 等利用一种聚合树脂 SEPABEAD EXE120 成功固定化西他列汀转氨酶( 突变型 ATA-117) ,固定化酶以异丙醇为溶剂,在 200 g·L- 1底物浓度下,重复使用 10次,连续反应 200 h,其活性并无损失。Mallin等利用优化的壳聚糖固定化两个 R-TA,两个固定化酶对于 1-苯乙胺的合成都有很好的活性,其中一种转氨酶转化在合成 R-2-己胺时,活性比游离酶提高了 13. 4 倍,转化率大于 99%。此外,Matosevic 等利用固定化酶微反应器平台成功实现了手性氨基醇多步生物转化筛选过程。由以上例子可以看出,转氨酶的固定化在提高酶稳定性、拓宽酶使用范围以及改善酶反应条件方面都有重要意义,通过固定化,有望克服游离酶反应的过程瓶颈,实现转氨酶工业化应用。但是固定化酶由于传质限制,酶活性会普遍降低,同时固定化酶的制备增加了酶的成本,因此更加优越的固定化介质有待开发。
3 转氨酶过程工程
转氨酶催化的转氨反应是一个热力学平衡过程,主要包括两个互补的反应: 手性胺的不对称合成和外消旋胺的运力学拆分。以上两个反应过程受反应物、产物、反应条件等因素影响,反应时所发生的平衡逆向移动是转氨反应中的瓶颈问题。因此,要实现高效率不对称合成就需要从影响因素入手,改变反应的平衡。众多学者就此问题进行了深入的研究,现将解决方案归纳如下:
3. 1 加入过量的氨基供体
改变平衡的一个最简单的方法就是加入过量的氨基供体,Savile 等在生产西他列汀时就采用了此方法: 在优化的反应条件下,向 40 mL 反应体系中加入2 - 丙胺39. 8 mmol,前西他列汀酮18. 9 mmol,此时氨基供体的量是氨基受体的2 倍; 在放大的反应体系中,加入高达 10 倍过量氨基供体,反应得到西他列汀的转化率为 92%。
需要注意的是此种方法只适用于反应的平衡对产物产率影响不大的情况。如果平衡严重不利于产物的生成,而反应所需的底物质量浓度比较高( >50 g·L- 1) ,氨基供体和受体按照 1 ~ 50∶ 1的摩尔比进行反应,氨基供体的过量倍数将会受到限制,加入太多的氨基供体将会出现可溶性差等其他的问题。
3. 2 副产物的自动降解
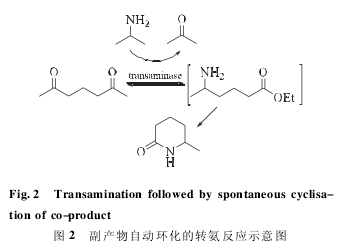
副产物自动降解是利用副产物在反应条件下自动形成另外一种非反应成分,达到解决抑制的目的。Lo 等发现以鸟氨酸和赖氨酸作为氨基供体时,反应产生的氨基酮会自动环化,使平衡向生成胺的方向移动。同样 Truppo 等在利用转氨酶合成 6-甲基-2-吡咯酮( 50 g·L- 1) 的反应中( 图 2) ,利用一级产物乙胺基丁酰乙酯的自动环化,形成非抑制性终产物 6-甲基-2-吡咯酮的反应趋势,解除一级产物抑制,获得终产物时转化率大于 90%,产物 ee 值大于 99%。副产物自动降解是一种非常传统的方法,受到副产物性质限制,其应用并不多。
3. 3 移除产物或副产物
由于产物或副产物的存在,常常会对平衡产生抑制作用,因此可以通过在反应时移除产物或者副产物的方法来改变平衡,这种方法也称为在位产物移除( in situ product removal,ISPR) ,ISPR的效率与产物胺以及其它反应组分的性质有关。
其中产物的物理化学性质如挥发性、溶解性、带电性、疏水性、和分子大小是 ISRP 中研究最多的几个方面。
实现 ISPR 的途径有多种。Koszeiewski 等在利用 ATA-117 转化 4-苯基-2-丁酮合成 R-4-苯基丁-2-胺的过程中,综合运用有机溶剂萃取和调整pH 的方法使反应物的转化率达到 92% ,ee 值高达 99%; Truppo 等在利用 ATA-113 和 ATA-117 转化苯乙酮合成 α-甲基苯乙胺的过程中采用树脂提取技术成功获得了高达 99% 的转化率,与之前不采用 ISPR 技术的转化率 10% 相比,反应的转化率得到了大幅度的提高。另外让易挥发的副产物挥发掉也可以作为改变平衡的一个方法。
当以 2-丙胺或者 2-丁胺作为氨基供体时,会产生副产物丙酮或者丁酮,由于丙酮和丁酮与其它反应物相比具有较低的沸点,因此可以通过减压使副产物挥发掉,这样转氨反应就趋于完全。此外对于易挥发的胺还可以通过蒸馏的方法进行产物回收,这种方法多用于动力学拆分。
3. 4 酶偶联反应
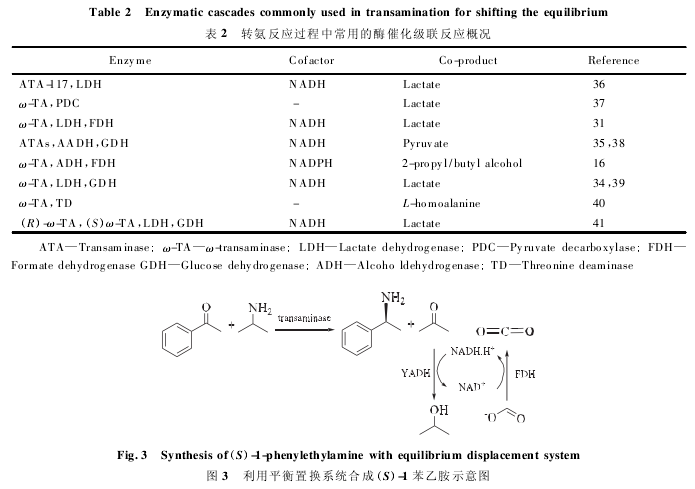
酶偶联反应是指将转氨反应与其它酶促反应联合使用,将副产物( 丙酮酸或者丙酮) 转化为非反应成分或者原始的底物。此方法是到目前为止研究和应用最多的提高不对称合成产率的途径,表 2 对常用的酶级联反应进行了总结。Cassimjee等用来自 A. citreus 的具有 S-选择性的 ω-转氨酶作为催化剂,以异丙胺为氨基供体,在催化苯乙酮合成 S-1-苯乙胺的反应( 图 3) 中引入平衡置换系统: YADH/FDH( yeast alcohol dehydrogenase/formate dehydrogenase) ,其中 YADH 将生成的丙酮转化为 2-羟基丙醇,FDH 再生辅因子 NADH( nicotinamide adenine dinucleotide plus hydro-gen) ,驱动反应正向进行,成功获得高达 99% 的产率和 99. 9% 的 ee( enantiomeric excess value)值,未引入平衡置换系统的反应转化率只有60% ~ 80% 。Schatzle 等利用七种 R-型转氨酶合成 R-胺时,引入 LDH/GDH( lactate dehydro-genase / glucose dehydrogenase) 系统,通过优化反应条件,使得产物产率高达 99%。其中 LDH 将丙酮酸转化为 2-羟基丙酮酸,GDH 用来再生辅因子 NADH,转氨酶联合 LDH/GDH 达到了高效合成手性胺的目的,此外在以上两个反应体系中,由于使用辅因子再生系统,在一定程度上降低了反应的经济成本。Truppo 等在转氨酶 ATA-117拆分外消旋苯乙胺的反应体系中加入 D-氨基酸氧化酶,实现了丙酮酸再生,同时反应的平衡也发生了移动,反应 1 h,转化率可达到 50%,( S) -1-苯乙胺 ee 值高于 90%,而之前反应 1 h 转化率只有 7. 5%,产物 ee 值只有 8%。由此可见利用酶偶联反应可以大大提高不对称合成以及动力学拆分的产率,对于小规模制备来说是一种高效可行的方法。
合理的酶反应系统是获得高转化率的保证。酶的性质、反应体系中各成分的相互作用、辅因子的加入量等都会影响反应效率,比如在 FDH 体系中加入高浓度的甲酸盐会影响其它酶的活性和稳定性。Santacoloma 等对酶级联反应相关问题做了详细的讨论。酶级联反应的效率虽然很高但是其经济成本很高,不适合大规模制备。一个替代途径就是用全细胞作为催化剂。利用生物技术的方法实现目标酶在宿主菌中共表达,利用全细胞进行催化。Yun 等利用可以共表达 ω-TA和乙酰乳酸合成酶的 Ecoli 全细胞作为催化剂,成功合成了 S-α-甲基苯胺,Bea 等利用含有ω-TA 和内源性氧化还原酶的重组毕赤酵母为催化剂,成功拆分 α-苯乙胺,ee > 99%,转化率达52. 2% 。此外,Wang 等开发了一种新的单酶级联反应体系,用 ω-TA 催化高效不对称合成手性胺,克服了多酶级联反应的成本高、酶不兼容等缺点,但是此方法目前并不能用于手性胺的大量生产。
4 转氨酶应用
转氨酶主要用于手性胺、手性氨基酸、手性氨基醇等的合成,这些手性化合物在制药工业、农业、化工业都有重要的作用,其常作为药品的活性成分或主要中间体被使用。目前,立体选择性的合成以上手性化合物主要是通过化学法实现,如大家熟知的不对称还原 Stiff 碱,但是化学法存在一些不足,如反应条件苛刻、运用有毒的过渡态金属催化剂等,有时候在单一的催化反应中得不到足够的立体选择性,引起了一系列的环境问题,产品也不符合药物要求。然而利用转氨酶催化制备相应的手性化合物反应条件温和、过程简单、立体选择性高,逐渐取代了一些化学步骤。利用酶法、化学-酶法合成含手性胺的活性药物以及小分子手性胺非常具有发展前景。下面从四个方面介绍转氨酶在合成手性化合物中的应用。
4. 1 合成手性胺
手性胺是合成许多手性药物的重要中间体,也是含氨基的光学纯药物的主要成分。神经类药物、心血管药物、抗高血压药物、抗感染药物及疫苗等都是以手性胺作为中间体; 而抗糖尿病新药Januvia 的主要成分西他列汀是 R-型丁二胺。
在手性胺的合成中两者同样重要。下面逐一介绍。
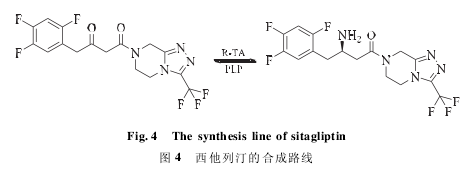
a. 不对称合成,是指在合适的氨基供体( 如异丙胺,L-丙氨酸) 存在下,转氨酶催化前手性酮生成相应的手性胺的过程。其又可细分为两类,酶一步合成法和化学 - 酶合成法。例如,Savile等利用突变型 R - 转氨酶 ATA - 117 不对称合成西他列汀,经过一步反应,成功得到西他列汀,实现了西他列汀 200 g·L- 1的工业化生产,24h 转化率 > 95% ,ee > 99. 5% ( 图 4) ; Fuchs 等成功采用化学酶法全合成得到( S) -利斯的明,其首先利用 ω 转氨酶合成所需的前体 1-苯乙胺,并利用酶偶联反应解决平衡移动问题,后利用化学法合成其余基团( 图 5) ,经过四步反应,整体收率达 71%,ee >99%。以上例子的成功都是非常激动人心的,但由于反应的可逆性,其转化率理论不能达到 100%,因此,解决平衡移动问题仍然是今后需要深入研究的。
b. 动力学拆分,是指在合适的氨基受体( 如苯乙酮) 存在下,转氨酶催化外消旋胺生成光学纯手性胺的过程。其实质也是转氨反应,只是在本反应中氨基供体为外消旋胺中一个构型的胺,转氨酶将一个构型的氨基转移至氨基受体上,得到与转氨酶选择性相反的光学纯的胺。理论上,转化率 >50%所得产物的 ee >99%,拆分效果较好。例如,Koszelewski 等利用 R 和 S-2 种转氨酶组成一锅两步法用于美西律的去消旋化,成功获得了光学纯的美西律,其收率达 97%,ee >99% ,这也是拆分和不对称合成相结合比较成功的例子( 图 6) 。动力学拆分能够成功得到高光学纯的产物,同时引入了底物酮,若采用 Koszelewski 所述的方法将能成功变废为宝,达到双赢的目的。
c. 工业应用情况: 一个生物催化剂要应用到工业 生 产 中,其 底 物 质 量 浓 度 至 少 应 达 到50 g·L- 1,因此大部分转氨酶,由于底物浓度、反应温度、有机溶剂耐受性等众多因素的限制目前只停留在实验室阶段。近几年转氨酶在工业应用最成功的例子当属突变型 ATA-117,其能够用于西他列汀的工业生产。除此外,其他小分子胺的生产也达到了工业要求,如 Truppo 等采用转氨酶与 LDH 偶联以及离子交换树脂在位移除产物两种方法,成功实现了 R/S 甲基苯乙胺以及 R/S 6-甲基-2-哌啶酮 50 g·L- 1的高效生产,其收率 > 90,ee > 99%。表 3 对近几年利用转氨酶生产手性胺的情况进行了归纳总结,由此可见转氨酶在制备手性胺方面具有较大的应用潜力和开发价值,是目前及未来的研究热点。
4. 2 合成手性氨基酸
光学纯的氨基酸在生物体内起着举足轻重的作用,同时在制药工业有着重要的作用。很多 α-和 β-氨基酸及其衍生物是生物体内关键的神经递质,如谷氨酸盐、γ-氨基丁酸; 而很多非蛋白氨基酸则在生物次级代谢过程中有重要作用,图 7对含氨基酸的药物进行了总结。2008 年之前,开发有药学活性的光学纯氨基酸吸引了众多药学家的眼球,利用转氨酶制备芳香族 β-氨基酸、L-α-氨基酸,脂肪族 L-α-氨基酸以及 D-氨基酸、15N 标记的氨基酸等层出不穷,Hohne 等对此进行了综述。
到目前为止,生物催化法制备手性氨基酸最成功的例子为 L-高苯丙氨酸的制备。L-高苯丙氨酸是制备血管紧张素转换酶( ACE) 抑制剂药物的重要原料,它是目前世界上约 20 种抗高血压新药的共同中间体,如依拉普利、地拉普利、西拉普利等。目前用于制备手性氨基酸的转氨酶种类很多如酪氨酸转氨酶、天冬氨酸转氨酶、ω - 转氨酶等。Lo 等用工程化的大肠杆菌天冬氨酸转氨酶高效制备了 L-高苯丙氨酸。Min 等用固定化的消旋酶及氨甲酰水解酶由相应底物制备L-高苯丙氨酸,此固定化酶可重复利用 14 次,实现了转氨酶的重复高效利用,更有利于工业化应用。
4. 3 合成手性氨基醇
手性氨基醇结构中含有手性氨和手性醇,是一类更有价值的生化试剂及药物中间体,如抗病毒糖苷酶抑制剂、鞘脂类药物,广谱抗生素氯霉素、甲砜霉素等众多药物中均含有手性氨基醇。目前生产手性氨基醇的方法有化学法和生物催化法 2 种,相对于化学法,生物催化法更加绿色、高效,因此便成为研究热点。
生物催化法制备手性氨基醇主要由有 3 种途径: a. 酮还原酶或转酮醇酶与转氨酶偶联,转化底物酮生成手性氨基醇; b. 转氨酶转化手性酮醇为手性氨基醇; c. 酮还原酶转化手性氨基酮为手性氨基醇。由此可见,转氨酶在手性氨基醇的合成中起着举足轻重的作用,利用转氨酶制备手性氨基醇也越来越受到化学家的亲睐。Smith等设计了一种简洁的合成手性氨基醇的方法: 转酮醇酶和 ω-转氨酶偶联,通过两步反应,成功制得( 2S,3S) -2-氨基戊烷-1,3-二醇,此法可以实现( 2S,3S) -2-氨基戊烷-1,3-二醇制备规模的生产。
Smithies 等利用 C. violaceum ω-转氨酶成功将1,3-二羟基-1-苯丙基-2-酮转化为( 2S) -2-氨基-1-苯基-1,3-丙二醇,而此酶对外消旋 1,3-二羟基-1-苯丙基-2-酮并无选择性( 图 8) ,因此其用于转化底物酮醇制备手性氨基醇非常理想。虽然以上例子均令人欣喜,但是能够高效制备手性氨基醇的转氨酶种类并不多,尤其是能用于工业化生产的更是少,因此,新的酶以及更优的反应条件亟待开发。
4. 4 合成含氘氚标记的手性胺
放射性氘、氚标记的化合物对于了解药物代谢和药物动力学有重要作用。氘氚标记的药物在临床前研究中,有利于研究者理解药物的吸收和分布,对提高其活性有重要指导意义。传统方法是从含氘氚标记的底物直接合成手性胺,而Truppo 等通过向反应体系中加入氘、氚的方法直接由前手性酮制备得到了多种重氢标记的手性胺,如含氘西他列汀的制备( 图 9) 。在 60% 的氘水和 40% DMSO 溶 剂 中,经 过 20 h 转 化,20 g·L- 1的底物有 82% 转化为含氘西他列汀,此西他列汀中所有氢均被置换为氘。利用转氨酶CDX-017 制备氚标记的 N-Cbz 吡咯胺,在优化的反应条件下,经过 2 d 反应,N-Cbz 吡咯胺中氚的富集程度达到 280%,明显高出统计值 100%。以上例子说明转氨酶同样可以用于催化前手性酮制备多种重氢标记的手性胺,其过程简单可操作性强,是今后的发展趋势。
5 结语
近年来,转氨酶在手性化合物合成中得到了广泛应用,其凭借绿色、高效的合成优势吸引了无数化学家的关注,人们采用化学 - 酶法结合的手段制备得到了大量手性胺。近五年来,从转氨酶蛋白工程、表达、固定化、进程工程到应用,国内外研究者都投入了大量的精力,也得到了很多可喜的成果。但是目前能够用于工业化生产的转氨酶并不多,因此新型酶的筛选以及反应条件的优化仍然是目前利用转氨酶合成手性化合物面临的两大挑战。开发新酶、改造现有酶以及研究转氨酶过程工程等工作仍将是研究者们今后的研究重点,作者希望开发出更多可用于工业化生产的转氨酶,以期搭建起手性胺的生物催化生产平台。
参考文献:
[1] 许良葵,林华庆,张蜀. 新手性药物左旋沙丁胺醇的研究进展[J]. 中南药学,2008,6( 1) : 85 -88.
[2] WARD J,WOHLGEMUTH R. High-Yield biocatalyt-ic amination reactions in organic synthesis[J]. CurrOrg Chem,2010,14( 17) : 1915.
[3] DESAI A A. Sitagliptin manufacture: A compellingtale of green chemistry,process intensification,and in-dustrial asymmetric catalysis[J]. Angew Chem IntEd,2011,50( 9) : 1974.
[4] BERGLUND P,WOODLEY J. 1st International sym-posium on transaminase biocatalysis [EB / OL ].( 2013 -02 -03) [2013 -02 -15]. http: / /www. bio-tech. kth. se / biochem / transam / .
[5] LUTZ S,BORNSCHEUER U T. Protein engineeringhandbook [M ]. Germany: Wiley Online Library,2009: 1 - 2.
[6] BORNSCHEUER U T,KAZLAUSKAS R J. Findingbetter protein engineering strategies[J]. Nat Chem Bi-ol,2009,5( 8) : 526.
[7] BEHRENS G A,HUMMEL A,PADHI S K,et al.Discovery and protein engineering of biocatalysts fororganic synthesis [J]. Adv Synth Catal,2011,353( 13) : 2191 -2215.
[8] BORNSCHEUER U T,HUISMAN G W,KAZLAUS-KAS R J,et al. Engineering the third wave of biocatal-ysis[J]. Nature,2012,485( 10) : 185 - 193.
[9] BOMMARIUS A S,BLUM J K,ABRAHAMSON MJ. Status of protein engineering for biocatalysts: how todesign an industrially useful biocatalyst[J]. Curr OpinChem Biol,2011,15( 2) : 194 - 196.
[10]SAVILE C K,JANEY J M,MUNDORFF E C,et al.Biocatalytic asymmetric synthesis of chiral aminesfrom ketones applied to sitagliptin manufacture[J].Science,2010,329( 5989) : 305 - 309.
[11]SVEDENDAHL M,BRANNEBY C,LINDBERG L,et al. Reversed enantiopreference of an ω-transaminaseby a single-point mutation [J]. Chem Cat Chem,2010,2( 8) : 976 - 980.
[12]CASSIMJEE K E,HUMBLE M S,LAND H,et al.Chromobacterium violaceum ω-transaminase variantTrp 60Cys shows increased specility for( S) -1-phenyl-ethylamine and 4'-substituted acetophenones and fol-lows Swain-Lupton parameterisation[J]. Org BiomolChem,2012,10( 28) : 5466 - 5470.
[13]HUMBLE M S,CASSIMJEE K E,ABEDI V,et al.Key amino acid residues for reversed or improved en-antiospecificity of an ω-transaminase[J]. Chem CatChem,2012,4( 8) : 1167 - 1172.
[14]ITO N,KAWANO S,HASEGAWA J,et al. Purifica-tion and characterization of a novel( S ) -enantioselec-tive transaminase from Pesudomonas fluorescensKNK08 - 18 for the synthesis of optically active a-mines[J]. Biosci Biotechnol Biochem,2011,75( 11) :2093 - 2098.
[15] IWASAKI A,MATSUMOTO K,HASEGAWA J. Anovel transaminase,( R) -amine: pyruvate aminotrans-ferase,from Arthrobacter sp. KNK168 ( FERMBP-5228) : purification,characterization,and gene cloning[J]. Appl Microbiol Biotechnol,2012,93( 4) : 1563 -1573.
[16] CASSIJEE K E,BRANNEBY C,ABEDI V,et al.Transaminations with isopropylamine: equilibrium dis-placement with yeast alcohol dehydrogenase coupledto in situ cofactor regeneration[J]. Chem Commun,2010,46( 30) : 5569 - 5571.
[17]BEA H S,SEO Y M,CHA M H,et al. Kinetic Reso-lution of α-methylbenzylamine by recombinant Pichiapastoris expressing ω-transaminase [J]. BiotechnolBioproc E,2010,15( 3) : 429 - 434.
[18]WEINHANDL K,WINKLER M,GLIEDER A,et al.A novel multi-enzymatic high throughput assay fortransaminase activity[J]. Tetrahedron,2012,68( 37) :7586 - 7590.
[19]KWON Y C,LEE K H,KIM H C,et al. Cloning-Inde-pendent expression and analysis of ω-transaminases byuse of a cell-free protein synthesis system[J]. ApplEnviron Microbiol,2010,76( 18) : 6295 - 6298.
[20] REHN G,GREY C,BRANNEBY C,et al. Activityand stability of different immobilized preparations ofrecombinant E. coli cells containing ω-transaminase[J]. Process Biochem,2012,47( 7) : 1129 -1134.
[21]FERN?NDEZ M C,NETO W,L?PEZ C,et al. Im-mobilization of Escherichia Coli containing ω-transam-inase activity in LentiKats[J]. Biotechnol Prog,2012,28( 3) : 693 - 698.
[22]TRUPPO M D,STROTMAN H,HUGHES G. Devel-opment of an immobilized transaminase capable of op-erating in organic solvent[J]. Chem Cat Chem,2012,4( 8) : 1071 - 1074.
[23]MALLIN H,MENYES U,VORHABEN T,et al. Im-mobilization of two ( R) -amine transaminases on anoptimized chitosan support for the enzymatic synthesisof optically pure amines[J]. Chem Cat Chem,2013,5( 2) : 588 -593.
[24]KOSZELEWSKI D,MULLER N,SCHRITWIESER LH,et al. Immobilization of ω-transaminases by encap-sulation in a sol-gel / celite matrix[J]. J Mol Catal B:Enzym,2010,63( 1 /2) : 39 - 44.
[25]CASSIMJEE K E,KOURIST R,LINDBERG D,et al.One-step enzyme extraction and immobilization forbiocatalysis applications [J]. Biotechnol J,2011,6( 4) : 463 -469.
[26]MATOSEVIC S,LYE G L,BAGANZ E. Immobilisedenzyme microreactor for screening of multi-step bio-conversions: characterisation of a de novo transketo-lase-ω-transaminase pathway to synthesise chiral ami-no alcohols[J]. J Biotechnol,2011,155 ( 3) : 320 -329.
[27] TUFVESSON P,RAMOS J L,JENSEN J S,et al.Process considerations for the asymmetric synthesis ofchiral amines using transaminases[J]. Biotechnol Bio-eng,2011,108( 7) : 1490.
[28]KOSZELEWSKI D,LAVANDERA I,CLAY D,et al.Asymmetric synthesis of optically pure pharmacologi-cally relevant amines employing ω-transaminases[J].Adv Synth Catal,2008,350( 17) : 2761 - 2766.
[29] H?HNE M,TOBINS K,BORNSCHEUER U T. Aprotection strategy substantially enhances rate and en-antioselectivity in ω-transaminase-catalyzed kineticresolutions[J]. Adv Synth Catal,2008,350 ( 6 ) :807 - 812.
[30]LO H H,HSU S K,LIN W D,et al. Asymmetricalsynthesis of L-homophenylalanine using engineeredEscherichia coli aspartate aminotransferase[J]. Bio-technol Prog,2005,21( 2) : 411 - 415.
[31]TRUPPO M D,ROZZELL D,TURNER N J. Efficientproduction of enantiomerically pure chiral amines atconcentrations of 50 g·L- 1using transaminases[J].Org Process Res Dev,2010,14( 1) : 234 - 237.
[32] LYE G J,WOODLEY J M. Application of in-situproduct remove techniques to biocatalytic processes[J]. Trends Biotechnol,1999,17( 10) : 395 -402.
[33]GEORGE M,MOHIT B,WEI LANG,et al. Enzymeand reaction engineering in biocatalysis: synthesis of( S ) -methoxyisopropylamine[J]. Chimia,1999,53( 12) : 584 -589.
[34]SCH?TZLE S,MUNSBERG F S,THONTOWI A,etal. Enzymatic asymmetric synthesis of enantiomerical-ly pure aliphatic,aromatic and arylaliphatic amineswith ( R ) -selective amine transaminases [J]. AdvSynth Catal,2011,353( 13) : 2439 - 2445.
[35]TRUPPO M D,TURNER N J,ROZZELL J D. Effi-cient kinetic resolution of racemic amines using atransaminase in combination with an amino acid oxi-dase[J]. Chem Commun,2009,16: 2127 - 2129.
[36]KOSZELEWSKI D,LAVANDERA I,CLAY D,et al.Formal asymmetric biocatalytic reductive amination[J]. Angew Chem Int Ed,2008,47 ( 48) : 9337 -9340.
[37] HOHNE M,KUHL S,TOBINS K,et al. Efficientasymmetric synthesis of chiral amines by combiningtransaminase and pyruvate decarboxylase[J]. ChemBiochem,2008,9( 3) : 363 - 365.
[38]TRUPPO M D,TOZZELL J D,MOORE J C. Rapidscreening and scale-up of transaminase cattalysed reac-tions[J]. Org Biomol Chem,2009,7( 2) : 395 - 398.
[39]FUCHS M,KOZELEWSKI D,TAUBER K,et al.Chemoenzymatic asymmetric total synthesis of ( S ) -Rivastigmine using ω-transaminases[J]. Chem Com-mun,2010,46( 30) : 5500 - 5502.
[40]MALIK M S,PARK E S,SHIN J S. ω-Transaminase-catalyzed kinetic resolution of chiral amines using L-threonine as an amino acceptor precursor[J]. GreenChem,2012,14( 8) : 2147 - 2140.
[41]KOSZELEWSKI D,CLAY D,ROZZELL D,et al.Deracemisation of α-chiral primary amines by a one-pot,two-step cascade reaction catalysed by ω-transam-inases[J]. Eur J Org Chem,2009,14: 2289 - 2292.
[42]SANTACOLOMA P A,SIN G,GERNAEY K V,etal. Multienzyme catalyzed processes: Next generationbiocatalysis [J]. Org Process Res Dev,2011,15( 3) :203 - 212.
[43]YUN H,KIM B G. Asymmetric synthesis of( S) -α-methylbenzylamine by recombinant Escherichia colico-expressing omega-transaminase and acetolactatesynthase [J]. Biosci Biotechnol,2008,72 ( 11 ) :3030 - 3033.
[44]WANG B,LAND H,BERGLUND P. An efficient sin-gle enzymatic cascade for asymmetric synthesis ofchiral amines catalyzed by ω-transaminase[J]. ChemCommun,2013,49( 2) : 161 - 163.
[45]NUGENT T C. Chiral amine synthesis: methods,de-velopments and applications[M]. West Sussex,UnitedKingdom: John Wiley & Sons,2010: 225 - 246.
[46] MALIK M S,PARK E S,SHIN J S. Features andtechnical applications of ω-transaminases [J]. ApplMicrobiol Biotechnol,2012,94( 5) : 1163 - 1171.
[47]KOSZELEWSKI D,PRESSNITZ D,CLAY D,et al.Deracemization of mexiletine biocatalyzed by ω-trans-aminases [J]. Org Lett,2009,11( 21) : 4810 - 4812.
[48]HANSON R L,DAVIS B L,CHEN Y,et al. Prepara-tion of( R) -amines from racemic amines with an( S) -amine transaminase from Bacillus megaterium [J].Adv Synth Catal,2008,350( 9) : 1367 - 1375.
[49]MUTTI F G,FUCHS C S,PRESSNITZ D,et al. Ste-reoselectivity of four ( R) -selective transaminases forthe asymmetric amination of ketones[J]. Adv SynthCatal,2011,353( 17) : 3227 - 3233.
[50]H?HNE M,BORNSCHEUER U T. Enzyme Catalysisin Organic Synthesis: Third Edition[M]. West Sus-sex,Uuited Kingdom: John Wiley & Sons,2012: 779 -811.
[51]AHMAD A L,OH P C,ABD SHUKOR S R. Sustain-able biocatalytic synthesis of L-homophenylalanine aspharmaceutical drug precursor[J]. Biotechnol Adv,2009,27( 3) : 286 - 296.
[52]MIN C Y,WEN H H,SUNG C L. Synthesis of L-ho-mophenylalanine with immobilized enzymes [J].Process Biochem,2010,45( 5) : 667.
[53]SMITHIES K,SMITH M E B,KAULMANN U,et al.Stereoselectivity of an ω-transaminase-mediated ami-nation of 1,3-dihydroxy-1-phenylpropane-2-one[J].Tetrahedron: Asymmetry,2009,20( 5) : 570 - 574.
[54]SMITH M E B,CHEN B H,HIBBERT E G,et al. Amultidisciplinary approach toward the rapid and repa-rative-scale biocatalytic synthesis of chiral amino alco-hols: a concise transketolase-/ ω-transaminase-media-ted synthesis of ( 2S,3S ) -2-aminopentane-1,3-diol[J]. Org Process Res Dev,2010,14( 1) : 99 -107.
[55]VOGES R,HEYS J R,MOENIUS T. Preparation ofcompounds labeled with tritium and carbon-14[M].West Sussex,United Kingdom: John Wiley & Sons,2009: 1 - 23.
[56]TRUPPO M,JANEY J M,GRAU B,et al. Asymmet-ric,biocatalytic labeled compound synthesis usingtransaminase [J]. Catal Sci Technol,2012,2 ( 8 ) :1556 - 1559.