
������ 2003 ��“�й����൰��������֯”(Chinahuman proteome organization, CNHUPO)�������� ,�й��ĵ�������ѧ�о�������ʮ���ķ�չ, ���ֳ��ټ��������ٻ���ŵľ���. ���й���ѧ������“ ������൰������ƻ� ”(human liver proteomeproject, HLPP)֮��, 2014 �� 6 ��, “�й����൰������ƻ�”(china human proteome project, CNHPP)�ھ�����, ��־���й���ѧ�ҿ�ʼ��ȫ�桢��ȷ�ز�������ȫ���ٵ������������߷���. ��������������[1~4]�Ļ�����, ��������൰������ƻ���2010~2013���й���������ѧ�����ķ�չΪ�����������.
����1�� ������൰������ƻ��ķ�չ��ɾ�
����2003 ��, �ɺظ�����������Ŷ�[5]�����“������൰��������ʼƻ�”��ʼʵʩ, �����й���ѧ���״����ι����ش���к�����Ŀ. ������, �й���������ѧ�о��Ŷ����к��������Ϲ���, ������ 3 ������ȡ���˽��Ե��½�չ(ͼ 1): ϵͳ�Ե�ע���൰���ʱ����͵�����������(����); ���γ�ȵػ��Ƹ��൰���ʵ���ϸ����λ�����������ͼ(��ͼ); ��������˴��ģ�ĸ��൰������ѧ�о����Ϻ����ݿ�(���൰��������֯�����⡢�����Ϳ�Դ�������ݿ�, ����).
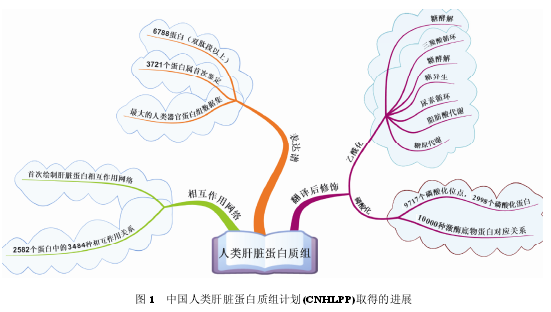
����1.1 ���൰�����������
��������������Ŵ����������滷�����������;���״̬�����������������, ����������������֯��������һ����������. Ϊ�˻��ƾ��д����Ե�������������, �й���������ѧ�Ŷ������ͬ�к���, ϵͳ�����˸�����֯�����������Ե�����ѧ�о������Ӱ��, �����˹�����������������֯���ٵ�������ѧ����Ʒ�Ʊ�������������(standard operating procedures, SOPs), Ϊ������൰��������ʼƻ���ʵʩ�춨�˻���[6]. �ڴ˻�����,�й���������ѧ�о��ŶӶԸ��������Ͳ���״̬�¸�����֯����������ϵͳ�ĵ�������ѧ�о�, ��������˫�Ķ����ϸ߿��ŵĸ��൰���� 6788 ��. ����3721 ���������ڸ�����֯�б��״μ���. ��������Ϊֹ���൰������ѧ�о��ƻ������ĵ�һ��֯�����ĵ����������ݼ�, ���첢�ٽ��˹��ʵ�����������ƻ������뿪չ.
�����й���������ѧ�о��Ŷӽ�������Щ���������ʵķ����Ϣ������ϵͳ�о�, ������Щ�����ʺ��6 ���������ķ�ȷ�Χ, ���� 78%�ĵ�����(5294 ��)λ���еȻ�ƫ���ź�ǿ������, ���״μ�����3721�������е�3069���������ڵͷ�ȵ���. ���ڸ����еͷ�ȱ����ϸ��ɫ��P450�����4�����Ӻ�3������ͨ����ص����ڸ�����֯�б���Ч����[7,8].
����1.2 ���൰�������������൰���ʵķ��������, �����ữ�����������о�Ҳ���㷺��չ. ������ѧ����������Ծ�Ŷ�[9]�ڸ��൰���ʵ���������������չ���г�Ч���о�, ��չ�����������Ͳ��������´�л������ص��о�����. ����ʵ�����Լ����Ƶ������Ч���������Ķθ�������, ʵ���˸�����֯�д��������������Ķεĸ����ʹ��ģ����. ����Щ�������������Ķν���ϵͳ��������Ϣѧ�о�, ���ּ������в����м��л��ø����, ���ǽͽ⡢��������������ѭ��������ѭ����֬�������ԭ�ϳɵ�;���ĵ����ʱ�����������. ��Щ��лø�����ʷ��ӵ����������γ̶���ϸ������������, �������ǡ��������֬�����Ũ�ȹ�ϵ����. ��Щ�����ʾ, ����ø�������������ζ�ϸ���ڵ�������л������Ҫ�ĵ�������.
�����������, �Թ�������������Ծ�Ŷ�[10]����, ��ɳ���Ͼ�(Salmonella)Ϊ�о�����, �����ڲ�̼ͬԴ����������, ���Ĵ�лø���ӵ�����������ˮƽ�������ҵIJ���, ����Ӧϸ��������������л����Ҫ. ��������ø�����������λ���������ǽͽ�/��������������ѭ��/��ȩ��ѭ���Ĵ�лת������. ��Щ�о�����֤ʵ�˻�����������лø����ӵ�������������ԭ�˺���������и߶ȱ���, ͬʱҲ����ø�������������β����˻����л���̵ĵ���, �춨�˵�������������Ϊ��л�����ߵĻ���. ��Щ�о�������ֱ��� Science ͬһ����. Ϊ���ù���������Ծ�ڵ����ʷ�������ε�������ѧ�о��еĽܳ�����, �й�����������֯(CNHUPO)�ڵڰ˽��й���������ѧ���(2013, ����)Ϊ��䷢��ѧ������.
�������������ữ����Ҫ�Ĺ����źŴ�������, ���벢�������ڶ���������. �й���ѧԺ������ѧ�����о��������ŶӺͻ��пƼ���ѧѦ���Ŷ�[11]����, ��չ�˴��ģ�Ļ��ڹ̶��������Ͳ�����(immobilized metal affinity chromatography, IMAC)�����ĸ��൰���������ữ�о�, ����չ�����ữ��������Ϣѧ����, �����˸�����֯�� 2998 �������ϵ� 9719 �����ữλ��. ����������ģ�����ữ�����ʼ������ݼ�, �������������ܰ��� 10000 ����ڵ�����ữ��ø�������Ե�����ӵ����ữ�����ʷ�������, Ϊ���ữ�ź�;����������ķ��ӻ����о��춨�˻���.
����1.3 ����������ú���ϸ����λ���繹�������������(protein-protein interaction, PPI)��Ϣ�Ľ�ʾ�����������˽⵰���ʷ���������ϸ���ڵķ��ӻ���, ���ҿɽ�һ��̽����Щ���ӿ��ܲ���Ĵ�л;�����ź�ͨ·, �Ӷ�Ϊ��Щ�����ʵĹ��ܼ�����ӻ����о���������. ��� PPI ����Ĺ���Ҳ�� HLPP �ƻ�����Ҫ�о����ݺ���ҪĿ��֮һ. ͨ�����ģ��������ѧ�о������Ѿ��γɶ���ģʽ���� , ����ֲ�ԭϸ�� �� ��ĸ (Saccharomycescerevisiae) �� �߳� (Caenorhabditis elegans) ��Ӭ(Drosophila melanogaster)�ȵĵ��������������ͼ. ��Щ����õķ���Ϊ��������ѧ���������ӻ����о���ҩ��ɸѡ�ṩ��������ʵ�������֧��.
������Ŀǰ PPI �Ľ�����Ҫ���ٵ���ս��������Ⱦ���Ĵ��ڴ����ļ�������ƫ��, ������ѧʵ���о�����������ٲ��ֿɱ�ʵ��֤ʵ.
�����������������о����ĺظ������������������Ŷ�[12]ѡȡ�˸�����֯�����е� 5026 �������ӽ�����ϵͳ����ĵ�����-����������ù�ϵ�о�. ���ó���Ľ�ĸ˫�ӽ�����ƽ̨���ϸ�ļ������ų�����, ���Ŷӳɹ��ؼ����� 2582 �������ʵ� 3484 �������. ͨ�����ﻯѧ���������ѧ��ϸ�����ں�ɸѡϵͳ��֤��������õ������ʸߴ� 72%. ����ط�����Щ���������, ���Ŷӻ�������ϵ�о����������������Լ�����״̬�Ķ��еĵ����������. ����������൰�������������(humanliver protein interaction network, HLPN)���ʺ�����Ŀ��������ɵ������������ٵ�������ô������ݼ�. ��������ϵͳ������������������൰������������繦�ܾ�����Ҫ�ļ�ֵ.
����������൰������ƻ�(HLPP)���о���չ�����ڵ�������ѧ�о������Ľ����͵�������ѧ�ķ�չ.HLPP �Թ���“���ס���ͼ������”Ϊ��Ŀ��, Ϊ��������ѧ�������з��������ȷ�Ŀ�ѧ����ͷ�չĿ��.
����2�� ��������ѧ�����ķ�չ
������������ѧ���߲�ε��о��벻�������ķ�չ,�������ķ�չ��Ϊ��������ѧ�ṩ�µ��ӽǺ�˼ά��ʽ. �ڹ�ȥ�� 3 ����, �й���������ѧ�о��Ŷ��ڵ�������ѧ��Ʒ�Ʊ�����������Ʒ�ĸ�Чɫ���롢��������ε����ʵĸ�����������������Ͷ��������Լ�������Ϣѧ���ߵķ�չ�ȵ�������ѧ�о��ļ������з��涼ȡ����������չ.
����2.1 ����������Ʒ���Ʊ�
������������ѧ��Ʒ��Դ��ϸ��������֯�ڲ���ȫ��������, ���е��������ิ�Ӷ�������ȷ�Χ�������ص�, �����������ĸ߸��Ǽ����߾��ȶ�������˾������. ����һЩʵ���Ҷ���Ʒ���Ʊ����ڽ����˴�����̽��, �����˶��ֻ�ѧ��������ʲ���, ��һ���̶��������˸߷�ȵ��Ը߸��ǵ�������ѧ������Ӱ��, �����˵�������ѧ�о���������.
����(1) �ͷ�ȵ����ʻ�ѧ�������ʵĿ���. �Դ�����Ϊ����ż�������ͽ���, �γ��˶��ָ�Ч�ĵ����ʷ���������ϲ���, ���Ѿ���Ϊ��ѧ���ʷ���������Ч�ķ���.
�������͵Ĵ��Բ��϶��Դ������������л��߷��ӽ���γɾ�������ṹ�Ĵ�����, �ڴ˹�����ͨ�����ۼ�������Եȷ�����������治ͬ�Ĺ��ܻ���. ������ѧ���Mԭ�ŶӺ��������Ŷ�[13]���ݽ���������ɫ��ԭ��, �� Cu2+���ӹ̶��ڸ߱ȱ�����Ľ����������������, ���ø���Ķ��1102�����Ժ��ܶȵ� Cu2+���ĶεĽ����, ���Խϸ�Ч�ش����Ļ����Ʒ�и����Ķ���Ʒ[13]. ��������[14,15]�ϳ��˾���Yolk-Shell�ṹ�Ĵ��Խ��̼�����. ���øò��ϵ��пṹ��ǿ������Ӧ, ���Դ���Ѫ����ѡ���Ե���ȡ�ͷ�ȵ���Դ���Ķ�. ���Ŷ������������20 ?L����Ѫ����Ʒ�и�Ч����ȡ���� 3402 �ֲ�ͬ����Դ�Զ���. ��Щ��Դ�Զ���ͨ����Ƚϵ�, ��������������. ���Ч����ΪѪ���е������־���ɸѡ����������. �й���ѧԺ������ѧ�����о�����������������Ŷ����Ͽ���ѧ�������Ŷ�[16,17]����, ͨ�������ѧ�ķ����� Fe3O4���������ǰ��������Ṳ������, �Ʊ�����ǿ���Ժͳ��������������Ͳ���. �ò��Ͽ�����������Ѫ�쵰��Ѫ���и߷�ȵ���. ��Щ�¼����ķ�չ���²��ϵĿ�����Ч��ȥ�����߽�����ѪҺ��Ʒ�и߷�ȵ��ĸ���, �������˵ͷ�ȵ��ļ������������и��Ƕ�, ����˴�ѪҺ��Ʒ�з��������־�������, ��˾������õ����ۺ�ʵ�ü�ֵ.
����(2) �����ͷ�ȵ����ʵ�������ʵ��з�. ����ض�������ѧ����, ��ƿ�����������Ե��������Ԫ��, ��ʵ������������ӵĸ�Ч����ĸ���.
�����������������о������ؾ��Ŷӷ�����һ��ת¼���� DNA ������д�������(catTFRE), �ܹ�����ϸ����Ʒ�и�Ч�ʵĸ���ת¼����. ���ø��ͽ��ʴӵ���ϸ����Ʒ�м������� 400 ���ת¼����, ���� 11 ����ͬ���͵�ϸ���й������� 878 ��ת¼����,������ϸ���ڽ� 1/2 �Ļ���������ת¼���Ӳ���,ʵ����ת¼���ӵĸ߸��Ǽ���. ͬʱ, �ÿ����������������[19]���úɶ��ɷ�ӦԪ��(HREs)DNA ������Ϊ�ͽ���, �ɹ��ش�С��(Mus musculus)������֯��Ʒ�и������˵ͷ�ȵ���Դ�Ժ���������. ������ʷ������������ڿ���Բ�ͬ������ѧ����, ��Ʋ�ͬ�ķ�Ӧԭ��, ����Եظ�Ч�����ͼ����ͷ�ȵĵ���������, ������ʾ������صĸ��ӷ��ӻ���. ��Ч����Ŀ�ĵ������о���Щ�����ʹ��ܵ�ǰ��. ��Щ��Ч�Ļ�ѧ���ʺ�������ʵĿ���, ��ʵ��Ŀ�ĵ���������ĸ���, ��Ч�������˵�������ѧ�IJ������, ��ǿ�����õ�������ѧ���������������ص�����.
����(3) ��������Ʒ��Чø�д����������о���չ.Ŀǰ, ��������ѧ�ļ����Ͷ�����Ҫ���ô��Ķε�����, �����¶���(bottom up)�IJ���, ��˵�����ˮ���Ϊ�ĶεĹ����Ǹ߶˵�������ѧ�����о��е���Ҫһ��. ���ʵ�ֵ���������Ʒø�����ĸ�Ч�����������ǵ�������ѧ��Ʒ�Ʊ������о����ȵ�.
����������ѧ���������[20]������һ��оƬ��Ӧ��, ͨ��������ø�̶��ھ��нϴ�ȱ����������������, ���̶ȵ�����˵���ø�͵�������Ʒ�ĽӴ����, ����˵��ﵰ���ʵ�ˮ��Ч��. ���ø÷�Ӧ��, �ȵ���øˮ�⵰���ʵ�����ٶȿɴﵽ 400mmol L-1min-1g-1, ��ʵ�ֵ���������Ʒ�Ŀ���ˮ��. ����, �÷�Ӧ�����ܹ��������Һ��ɫ���������õ�������Ʒ���ƽ̨���кܺõļ�����, ��ʵ�ֵ�����ˮ����Ķε������.
����������ѧ�������ŶӺ����Mԭ�Ŷ�[21,22]���ּ��⸨��ˮ�����ߵ�����ˮ���Ч��, ����ˮ�ⷴӦ��ʱ��. �÷�����������, �����ڼ�������ʵ�ֵ��� 2 L ��Ʒ�Ŀ��١���Ч��ø��Ϳ��ټ��, ������ڸ�ͨ����������ѧ���о�[22]. Ϊ����ߵ�������ĸ��Ƕ�, ��������[23]���ö���ø��ϴ���ˮ��IJ���, ȡ�������õ�Ч��. �������������������[24]�����������������ߵ��Զ��������ʿ���ˮ�⼼����װ��, �ٽ��˸�ͨ��������ѧ�о�.
������Ȥ����, ��������[25]�����ȵ���ø��������ˮ��ø������, ��������ø�Ļ���. �����������״α���õ���ø��������ø�Ļ���. ���Ŷӷ��ֵ���ø��ˮ�������¾���ˮ��ø�Ļ���, ���ں��нϸ��л���Ļ�������������ø������. ��ø�ٱ�Ƿ�Ӧ�����º�, �������������Ը�, ����Ч���ٳ��滯ѧ��Ƿ������Ķν������Ӧ�ķ���. �ݴ�, ���Ŷӷ�չ������� N ���ȶ�ͬλ�ر���Ķε��·���, ���ɹ�Ӧ���ڶ�����������ѧ����. N �������Ա�ǵ��Ķο����γɷḻ�� b �� y ����, ��Ϊ�Ķδ�ͷ�������Ŀ����춨�����õĻ���.
����2.2 ����������������ķ�չ������ɫ��ϵͳ�ĸ�Ч���뼼���ķ�չ�Լ����١��߾��ȸ߷ֱ����������Ŀ���, ����ɫ��-�������ü���ƽ̨�ĵ�������ѧ��ȸ��dz�Ϊ��ʵ. ��άҺ��ɫ�����������������Ǵ��ģ���߸��ǵ�������ѧ�о�����Ҫ�ֶ�. �������������о����Ķ�衵���[26]������һ���١���Ч�ļ����Ͷ�������������о�����, ʹ�ü����Լ���������Ⱥ�Ч�ʵõ���˫�����. �������ַ���, �� 12 h��������ʱ���ڴﵽ 8000 ������ϸ���������ļ�����. ����“Fast-seq”����ʡ����“����”�������, ����ͨ���ڳ�����ޱ궨���ͻ��ڱ�Ǽ�������Ժ;��Զ����������鶨������. ͬ�����ô�ͳ�� 2D-MS ����ƽ̨, �Ը��൰������ѧ�о�ȡ���˲����ijɼ�[27,28].
�������Mԭ�Ŷ�[27]����ܶ��ݶ����ĺ� 2D-MS ����ϵͳ������ C57BL/6J С�����˵�������, ��Ч������748 ���ͷ�ȵ���, Ϊ����˵��������о��ṩ�˽��. ��άҺ��ɫ���뼼��Ҳͬ�����������ữ��������ѧ�о�. �����Ŷ�[29]������һ���µķ���-����(reverse phase-reverse phase, RP-RP)��άɫ�����ü���, ���е�һάѡ�����߸� pH ����Һ��ɫ����, �ڶ�ά�����ߵĵ� pH ����Һ��ɫ��, ��ֱ�Ӵ���������. ����ά���뼼�����漫��ؽ����˵�������Ʒ�ĸ�����, �ɹ��ؽ����ữ�Ķκ�λ��������������� 8000 ���. ����֮��, ��������ѧ�������־��ļ����뷢���Լ�ҩ����Ч�����ȷ��淢���Ŷ��ص�����[30~32]. Ѫ�����ڵ�������ѧ�о�������֧���������־���, ���������������Ƚ�������ϸ���������ײ���(hepatitis B virus, HBV)��ظΰ�ϸ�����ڵ�������, �������� 1365 ������,��һ�α�����MMP1(Metalloproteinase 1)�����ڳ��õ�������־���̥����(a-fetoprotein, AFP)���и��õ���������������[30].
����2.3 ������������ѧ����Ӧ��
�����ɶ��Ե������ǵ�������ѧ��չ�ı�Ȼ����.
�����Ե����������ȷ����Ч�Ķ����о�������شٽ�������Ե�����Ϊ�����������Ͳ������ӻ��Ƶ�����. ������, �������Ͷ�����Ǽ���������, ����������ѧ�о�����õ��˹㷺��Ӧ��. ��Щ����������Ʒ�Ʊ�ʱ������, ��Ϊ�ޱ궨������ѧ��Ƕ����ʹ�л��Ƕ����� 3 ����Ҫ�ļ�������. ���߸�����ȱ��, �������ߵĶ������Ƚϸ�, �ڶ�����������ѧ�о��еõ�����Ч���ƹ�.
����(1) ��ѧ��Ƿ��������Եķ�չ. ��ѧ��Ƿ�����������Ʊ��ĵ����ʻ������Ʒ���б�ǩ���,�ɶ��κ�ϸ������֯�����ٵȼ������еĵ���������Ʒ���ж����о�, �������, Ӧ�÷�Χ��. ����ͳ�ļ�������һ���ļ�������, �д������о��Ľ�. ���ͬλ����Ա������Զ�������(isobaric tags forrelative and absolute quantitation, iTRAQ)�ڸ��������������ڱ��ƫ��, �й���ѧԺ�����������о�����˹���Ŷ�[33]�������µ�ͬλ�ر�Dz���——�ͬλ�ذ�����Ӧ��ǩ��Dz���(deuterium isobaric aminereactive tag, DiART), ���������ֱ�Ƿ��������ѻ��ơ��ɶ����������Լ��������ȷ���������ʵ�ĶԱ�.
�����������, DiART �� iTRAQ �����еı��������ź�ǿ�ȸ�, �������ȸ�Ϊȷ, �������ֳ��� iTRAQ���͵ı������Ӷ���������ѹ��, �����������Ķβ���ľ�ȷ�����о�.
������������ζ�����������ѧ���߸������κ�ȷ����˫����ս, �Ѷȸ���. �����Ŷ�[34]��չ��һ���µ����ữ��������ѧ��������, �ܹ�������ߵ����ʶ������Ⱥ�ͨ��. �������顢ʵ�����һ���ظ�������ֱ���“��”��“��”��“��”�ȶ���ͬλ�ر�ǵĶ�����������֯���ΰ�ϸ�������ữ�Ľ������ر��, �����÷���ɫ��/ǿ���ӽ���ɫ��/����ɫ�����߶�άҺ��ɫ�Ա�Ǻ��ϵ���Ʒ������ȷ����, ���� 42 h �ķ��������ʱ, ʵ���� 00�����ữ�ĶεĽ� 2000 �����ữλ��ļ����Ͷ���[35].
�����ڶ�����ǩ����, �����Ŷ�[36]Ҳȡ���˽Ϻõijɹ�. ��С�鿪������ӱ���Ķ� N-��ͬλ�ر�Dz���, �ò���ʹ�ú��в�ͬ�ȶ�ͬλ�صļ�ȩ��Ϊ����Լ�, �������Ķε� N-�˷���������������,�Ӷ����б�Ƕ���. �ò����ڸ������������п��Դﵽ 98%�� N-��λ��ѡ�������Ժ� 99%�ı��Ч��.
����(2) ��л��Ƿ��������Եķ�չ. �ȶ�ͬλ�ش�л��Ǽ���(stable isotope labeling by amino acidsin cell culture, SILAC)�Ƕ�����������ѧ�еĽ��.
�������Mԭ����[35]����ʹ��13C6-Arg ��13C6-Lys ��������Ʒ�����ȶ���ͬλ�ش�л���. ����Ʒ�ȱ�����Ϻ�, ʹ�������Եĵ�������ø Lys-N �� Arg-C ��������, ������LysΪN-�˺�ArgΪC-�˵��Ķ�. �Է���ʼ���߽�β����Ʒ�����Ķβ���, ������Ʒ��һ�������з������ӵ� m/z ֵһ��, ��Щĸ���������Ѻ����ɵĶ���������ͼ�н�������ͬ����Ǩ�Ƶ�λ��b�� y ϵ�����Ӷ�. ���� b �� y ϵ�����ӶԵ��ź�ǿ�ȱ�, ��ʵ�ָ�Ϊ��ȷ�Ķ���. ������ N�˻� C�˵��Ķ�, �ܿ���һ��������ͼ���γ�һ��������λǨ�Ƶ����Ӷ�, ����������������Щĩ���Ķ�, ���ҿ�ʵ����Щĩ�����ӶԵ�ֱ�Ӷ���.
�����й���ѧԺ�Ϻ�������ѧ�о�Ժ�������Ŷ�[37]���� SILAC �Ķ�����������ѧ������ת��ҽѧӦ���ƽ���һ��. �ÿ����齫SILACС��Ӧ�õ�IgA��������Ѫ�����Һ�ĵ�������ѧ�о���, ������һϵ�п��ṩ�ٴ���ϵĺ�ѡ�ؼ�������, ��: ���� C3, ����, VDBP, ApoA1, IGFBP7 ��. ����SILAC ���С�������༲��С��ģ�͵���Һ��������ѧ��ֱ��Ӧ��, ��Ϊ�µ������־��ɸѡ�ṩ���µ�ϵͳ��������.
�������˶�������������Ʒ��, �й���ѧԺˮ�������о�������Ŷ�[38,39]�� SILAC �����ɹ�Ӧ�õ�microRNA(miR)���﹦�ܵĶ����о�, ѡ�� siRNA �����õ� U266 ������ϸ����Դ�� miR-21 ��ϸ��ϵ��Ϊ�о�����, Ȼ������ SILAC ������ miR-21 DZ�ڵİб���ӽ���ϵͳ�Ķ����о�, ����ʹ�� Westernblot �ͱ���������֤ʵ�� STAT 3 �������Ʒ���(protein inhibitor of activated STAT 3, PIAS3)�ܹ������ź�ת���Ӻ�ת¼������ 3 (signal transducer andactivator of transcription 3, STAT3)�ļ���Ĺؼ������� miR-21 ��ֱ�ӵ���.
����2.4 �������鷭��������о��ļ�����չ�����ʷ�������� (post translational modif-ication, PTM)�ڸ�����������ж���������Ҫ������.
��������Ŀǰ�о��Ƚ���������ữ���ǻ��������ػ�������, �ڵ����ʹ��ܺͶ�λ�ȷ��涼��������Ҫ������. �����ĵ����ʷ�������μ���������˵�������ĸ��ӶȺͶ�̬��Χ. ͬʱ���ڸ߷�ȵ�������ЧӦ, ʹ�öԵ����ʷ�������εļ����Ͷ�����Ϊ��ս. ���, ��չ��Ч�� PTM �������Զ����ļ��������Ե���Ϊ��Ҫ.
����(1) ���ữ��������ѧ�ļ�����չ. ���������ữ������Ŀǰ�о���Ϊ����ķ������������.
�����й���������ѧ�������ữ�Ķθ��������Ŀ�������������������Ϣѧ֧�ź����о��������ȡ����ͻ���ijɼ��ͽ�չ.
�������������ϵĿ���Ϊ��Ч����IJ����ữ�Ķ��ṩ�˱���. ��������ѧ�Ҵ������ӡ�������ϡ��ṹ��������г���, ��չ�˶������Ͳ���[40~43], �ܺõظ��������ữ�Ķεĸ���Ч�ʺ�������, Ϊ�������ģ�о����ữ���������ṩ�˶��ֺ�ѡ����.
�������ữ����������ϴ����������� MALDI-TOF ������������, ���з���֮ǰ��Ҫ���е���pH ֵ�����εȷ����IJ���. ��������[44]��ǿ�����ӽ���ëϸ��Һ��ɫ���� MALDI-TOF ��������, ���˽���������ǰ��Ʒ��������, ͬʱ��������Ʒ��ʧ. �÷����ʺ��ڵ�һ���ữ�������ữλ��ļ���, �ܹ������� 10~50 amol �����ữ�Ķ�.
���������ữ�Ķθ���������, �����ữ���ε��Ķεĸ����ͼ��������ѵ�. Ϊ�����⸻�����������ữ�Ķεľ�����ϵ, ���ɵ���[45]��ϸ�����˽�ϵ� Ti ���ӵ����ữ�Ķεı���, ����˸���Ч��.
����ͬʱ���ֵ� TiO2����������ʱ, ���������ڸ��������ữ�Ķ�. ����, ������ѧ�ͽ�������[46]�����������Ũ�ȶ��ڵ��������ữ�Ķ��� TiO2���ӵĽ�������й�ϵ, ���Ŷӷ�չ�˼��������ķ���, ͨ���ı��������Ũ�������ֶ����ữ�͵����ữ����.
����Ϊ��������ữ��������ĸ��Ƕ�, ��������[23]ͨ��������ϲ�ͬø�Ե�����ø�����Ʊ���Ʒ. ��� Glu-C �� Trypsin �Ե���������Ʒ�������������ữ�Ķθ���, ��С��� HeLa ϸ��ϵ�й������� 8062 ���������ữ�Ķε� 8507 �����ữλ��, ����ڵ�һ Trypsin ø��, ����������˽� 1 ��.
�������ӵĵ���������Ʒ�ķ����Ҳ���������ữ�Ķεĸ����ͼ���. �����Ŷ�[29]��չ�����ö�άҺ��ɫ�IJ��Խ������ữ�Ķεķ�����������ĸ�Ч������ϵ. �������ϵ��, �����������ߵĸ� pH ����ɫ�����Ķη���, Ȼ�����õ� pH Һ��ɫ�����߷��벢����������, �ɴ˿���� 30%�����ữ�Ķεļ�����. ͨ����ͬ���ữ�Ķθ������Ե����ͬ���ܹ���߸���Ч��, �ҽ����һ�������ڵĸ���ƫ�Ե�����. ���ɵ���[47]��չ��1������ǿ�����ӽ������ TiO2�����IJ���, ��һ������ǿ�����ӽ������������ữ�Ķ�, �����岿�ֲ���TiO2�������ữ�Ķ�. ����ϲ���ʵ���˶���Ʒ���ữ��������ĸ�Ч����ƫ�Եĸ�������.
�������˶����о�, ��������εĶ����о����ڽ�ʾ������ѧ���������Ҫ. ��������[48]����Ti(4+)-EPO ������Ϊ������, ����ͬλ�ر���˵���Դ�����ữ�Ķ�. ����չ�� pseudo-triplex �ȶ���ͬλ��˫�������, ʵ���˸�ȷ�ԡ���ͨ���Ķ������ữ�����������. ��Щ�����Ŀ���Ϊ����������չ���������ữ������������־��ķ��ֺͷ��ӻ����о�����������[34].
����(2) �ǵ�������ѧ�ļ�����չ. �����ʵ��ǻ������μ�ʧ������������״̬�Ͷ��ּ���������չ�����а�������Ҫ�Ľ�ɫ. �춬����(N)-���ӵ��ǵ��������о��Ѿ��γ��˽�Ϊ����ļ�����ϵ, ���õ��˱Ƚ�������о�. Ϊ�˸��� N ���ε�����, �����ŶӺ�����ѧ½�����Ŷ�[49~52]�����˻��ڴ����������ϵ�ëϸ������, ʵ���� N-�������ĵĸ�Ч����. Ϊ�˸��Ӹ�Ч�ط��� N-�������εĵ�������,�ܶ�ʵ����ϵͳ�о�����չ����Ӧ�ĵ�������ѧƽ̨, ʵ���˶�Ѫ���Լ���֯��Ʒ���ǵ�������ķ���[53~55]. ���������ǻ��ǵ������ǻ����е���һ����Ҫ��������ʽ, �������о�����. �������������о����ĵ�ǮС���Ŷ�[56]�ڴ��ģ�������������ǻ������ʵĻ����Ϸ�չ�˻��ڶ༶��Ӧ���Ķ�����������ѧ��������, ʵ���˶Ժ��������ǻ�����������λ��Ķ�����;�ȷ����.
���������ǵ���������������λ����, �����ǵ������ϵĶ���ͬ���ɱ���ϸ����, ������Ϊ�����־��. ǮС�����[57]��չ��һ���� PCGO(1-pyrenebutyryl chloride functionalized free grapheneoxide)ʵ�ֿ��١���Ч�Ķ�����ո�������, �ɱȽϼ��ط�����Щ�������. �����Ŷ�[58]ͬ����չ�˻��� OMC�� N-���յĸ�������, �÷���������Чȥ�������ʵ�ͬʱ�ֿ������Եĸ�������, ���������������ź�ǿ��. ���Mԭ����[59]��չ�˻���ø�й�������N-��ĩ�����������յķ���, ʵ���˶����������Ķ����Ƚ�. ��Щ�о�������ƶ����ǵ�������ѧ�������־����о�.
����(3) ���ػ���������ѧ�о������ķ�չ. ���ػ������dz����ĵ����ʷ��������֮һ. ���������ε�������Ҳ�ǵ����ʵ���������Է��ػ����ε�һ����ʽ. ��ͬ�ķ���������Я����ͬ�Ļ�ѧ�ṹ��Ϣ�����ݵ��ﵰ�����������ѧ����. ���ػ����ε���������Ҫ�ɷ�������ø(E3s)������. Ȼ��, ������������ػ����εĵ��ﵰ�������Է�������ø�в�Ϊ������֪. �й�ҽѧ��ѧԺ���Ѻ���[60]������һ���� E3 �뵰���ʽ�Ͻṹ������õĵ�������ѧ���������Լ��� E3 �ĵ��ﵰ�IJ���, ����ͨ�����ⷴӦʵ����֤�˸ü������Ե���Ч��.
��������һ�ָ�ͨ�����ض� E3 ø���������ε����ɸѡ����. ͨ������ E3 �뵰���ʽ�Ͻṹ������õĵ�������ɸѡ�������ɼ����ض����������εĵ���,���ֲ�֤ʵ�˷�������ø(E3 ubiquitin-protein ligase,LNX1) �鵼�� PDZ ���Ӽ�ø (PDZ-binding kinase,PBK)�����ػ�����;��, �Ӷ�����ϸ������ֳ, ����ϸ����ù�ص�������.
�����й���ѧԺ�Ϻ�������ѧ�о�Ժ�����Ŷ�[61]���û������ĵ����ʷ�������μ�������, ������Smurf1 �ܽ鵼�ᵰ��(axin)K29 λ�Ķ�۷��ػ�����.��һ���о��������� K29 �Ķ�۷����������鵼�ᵰ������-����ø��ϵ�Ľ���, �������������ε��ᵰ��ͨ���� Wnt ������ LRP5/6 ���������, ���� LRP5/6 �����ữ����, �������� Wnt/b-catenin �ź�ͨ·.
����(4) ��������������ѧ�����о���չ. ��������������������һ��ͬ��������Ҫ���﹦�ܵĵ����ʷ����������ʽ. ������������Ⱦɫ�����ܡ�ת¼���Ӽ����Լ����ڴ�л������ø�Ļ��Ե����������Ҫ����. �о����������������κ�Ĺ��ܺͻ��Ƶ�ǰ���Ǽ�������������������λ���. Ȼ��,ϸ���ڴ��ڴ����ĸ߷�ȵ����������ε��鵰��,��Щ�������鵰�Ĵ��ڽ�����ĸ��Ŷ��ܶ��������������鵰�ĸ�����������. ���, �������������ε��о���Ҫ���������ϸ���ڵͷ�ȵ��������Ķε��ʹ��������ķ�չ.
����������ѧ�������Ŷ�[62]����˿���������ϸ���ڵͷ���������Ķθ������·���. ���Ŷ����Ȼ���˷������������Ķε������Կ������Ծ����������ѽ�ø�� 288 λ�����ᷢ�����������ε������Կ���; Ȼ��ͨ����ϸ����ֵķ���, �����������鵰�ĸ߷��; ���, ����������������εĿ����������, ���LC-MS/MS����, �������������ε��Ķκ�λ��.
����2.5 ������Ϣѧ��չ
�������Ÿ�ͨ����������ѧ�������Ŀ��ٷ�չ,�������������ر��Ǹ߾����������ݵ�“��ըʽ”ӿ��, �����ݵĹ������������ھ��������ս. Ϊ��,����������Ϣѧ���������������ơ��Ķκ͵����ʵļ����Ͷ�������ͷ������������ע�͡���������ε����ʵļ����͵����ʽ����������㷨���������߷�չ�ȷ��棬����չ��ϵͳ������о�������ȡ����һϵ�н�չ. �������ݵ�����ھ�ʹ��������ѧ��Ϊ�µ��������η��ֵ���������. ����, �����������Ԥ�⡢������ motif ���ݿ�ͻ��ڵ����ʵ������־�����ݿ�ȿ���Ҳȡ���˽Ϻõ��о���չ.
����(1) �����ʼ�����������ص�������Ϣ���߿���.
���������������ݵĵ����ʼ������̰���ʵ��ͼ���������. ���㲿��һ�����ͼ��Ԥ���������ݿ��������������� 3 ������.
������ͼ��Ԥ��������, ��ȷ��ĸ����������ѡ�ܹ����ͼ����ļ�����, ���ͼ�����ʺͶ���ȷ��. �й���ѧԺ���㼼���о�����˼���Ŷ�[63]������ pParse, ����ͬλ�ط���е�һͬλ�ط����߷��λ�ù�ϵ��ѡ��ѡ���, �����������Ժ�ɫ��ǿ��ȷ����һͬλ�ط�, ʶ�����ֹ�ϴ���Ķ�, ��Ч�������ͼ������.
���������ݵ��������Ʒ���, �������������о���������ƽ�Ŷ�[64]�������Ķμ���������������PepDistiller, ���� MASCOT �ѿ�����������������. �����ҵ�ڹ㷺ʹ�õ��ʿ����� MASCOTPercolator[65], ���ø���ѿ����ʿص�����������������. PepDistiller ���õĶ��̼߳���Ҳ�ܴ��ӿ��ͨ���������ݵĴ����ٶ�. �����Ŷ�[66]�������ʿع��� BuildSummary, ͨ����ͼ������������ض����Է��������, ���Ե�����ˮƽ��������(false discovery rate, FDR)Ϊ���չ�������, ʵ���˶�������������������������, Ҳ�ʺ����Բ�ͬ��Ʒ���������͵IJ�ͬ���ݼ�������.
���������������������ݽ����Ļ�������. ��˼���Ŷ�[67,68]��������Ŭ��, �������ҹ�������ȫ����֪ʶ��Ȩ���������� pFind, ����ҵ�ڵõ��˹㷺�ƹ�. �й���ѧԺ�Ϻ�������ѧ�о�Ժ����ѧ�Ŷ�[69]Ҳ�����˻���֧���������Ķ�������������, ����һ��ͼ����, �������Ⱥ�ȷ�Զ�Ҫ����������������� MASCOT, ProFound ��.
������ͷ���������������ݿ��ֱ������ͼ����Ϣ�����Ķ�, ���������µ��������εļ���. ����ͷ�����㷨��ͼ�������ϸ�, �����˸��㷨�ڵ;����������ݵ�ʵ��Ӧ��. �о�����, �ܱ����ݿ�����������ͼ����ֻ�в�����һ���ͼ�ɱ���ͷ�����㷨��ȷ����[70]. ������ײ�յ�����(higher-energycollisional dissociation, HCD) �͵���ת������(electron transfer dissociation, ETD)���������ɵĸ߾��ȶ���������ͼ�Ķ������ѷ������ӵ������Ժá��������Ͷࡢ�������ȸ�, ���ʹ�ô�ͷ�����㷨������н���, ��ȡ�ýϺõ�Ч��. �� HCD �� ETD ͼ���������ӵ����ͻ����л�����, �ɽ�һ�������߾���������ͼ�Ĵ�ͷ����Ľ�����. �й���ѧԺ���㼼���о�����˼���ŶӺͱ���������ѧ�о����������Ŷ�[71]���������˴�ͷ�������� pNovo, ��������� HCD ͼ�������Ӹ߾����Լ����зḻ��internal ���Ӻ� immonium ���ӵ��ص�, ʹ�� pNovo��ȷ����ͼ�������dz������ݿ������������� 80%����, ����ͬʱ��Ч�����������������κͰ�����ͻ��. �ڸ����������� pNovo+��, �� HCD ͼ����,����ͬһ�Ķε� ETD ͼ�Ķ���������Ϣ(���� c, z���Ӽ�����������)����������ͼ�ڵ㹹���Ͷ�̬�滮�㷨��, ʹ�ó����ѿ�Լ 95%�ļ�������ɱ�pNovo+����[72]. pNovo ���µ���������õ��˺ܺõ�Ӧ��, �����й���ѧԺ���������о�������������[73]���� pNovo �������� 1 ��˿���ᵰ��øAs_TRY-5 ������������ As_SRP-1. �� 2 �ֵ��������߳�������е��ھ��ӻ���Ӿ������ƵĹؼ�����.
��������, ��˼���ŶӺͶ������Ŷ�[74]���������������Ķν��������������� pLink, ʵ���˹�ģ���Ļ�ѧ���������ʵĽṹ����. pLink ͨ��ͼ��Ԥ���ˡ���ѡ�����Ķγ�ɸ�� KSDP ͼ��ƥ���ֵ��Ż�[75], ʵ���˽����ĶεĿ��ټ���, ������������ڽ����Ķμ����� target-decoy ����, ��Ч�ؿ����˽��������� FDR ˮƽ. pLink ����Ч�Խ�һ���ڴ����ĵ�������Ʒ�������ʸ�������߹�������Ʒ��ģʽ�����ȫϸ���ѽ�Һ�ϵõ�����֤.
����������������ѧ�����Ƿ���Ҫ�ȶ�ͬλ�ر�ǿ��Է�Ϊ�ޱ궨�����б궨��������. �ڵ������ޱ궨������, ����ƽ�Ŷ��������ѧ������ѧл�����Ŷ�[76]�����������ޱ궨������LFQuant, ʵ�����ޱ�ʵ������µĵ����ʾ�ȷ����. LFQuant �������µĽ��������㷨, ����˼��㸴�Ӷ�, ������������Ч��, ʵ���˶Զ��ָ�ʽ�������ݵĶ�������. ������㷨�ڶ�������ݼ��Ͻ����˲��ԺͱȽ�, ����Ŀǰ�Ķ������� MaxQuant[77]�Լ�IDEAL-Q[78], Ŀǰ�Ѿ��ɹ�Ӧ�����й�����Ⱦɫ��ƻ��ȴ��ģ���ݵĶ���������[79]. ��Ի����ȶ�ͬλ�ر�����ݵĶ�������, ����ƽ��л�����Ŷ�[80]�������������б궨������ SILVER. �����������������, SILVER �ж���������µĶ����ɿ�������ָ����˴��, �ڲ����Ͷ��������Ե�ǰ����, ����˶���ȷ��. ��Ŀǰ���õĶ������� MaxQuant�� Proteome Discoverer ���, SILVER �����ܹ���SILAC���ȱ�Ƿ�ʽ���о�ȷ����, ���ܹ�����֧��15N ��ǵĶ������ݷ���, �������������ݶ���������ʹ�÷�Χ. ����, LFQuant �� SILVER �������û��ѺõĽ���, �ܹ����ж��������ϵͳչʾ��ͳ��ѧ����, �����û�ʹ��. ��˼���ŶӺͶ������Ŷ�[81]Ҳ�����Ƴ����б궨������ pQuant, ���Ķζ���ʱ���⿼���˲���ȫ��ǵ����, ͬʱ�ڵ����ʶ���ֵ����ʱ�����˷Dz���ģ�ͽ�������, ��Ч����䶨��ȷ��.
����(2) �����ʻ�����ѧ�ڻ�������ע���ϵ�Ӧ�ã�
�����������ʱ��, ��������ѧ�������������������ݿ���е�������ļ����Ͷ���, Ҳ���Է����������еĻ���ṹע�ͽ�����֤������. ԭ�����������С, ����ṹ��, ��ֱ��ͨ�������������л�������ע�ͺ�����. �������������Ӵ�, ����ṹ����, �������ע�������㷨�Լ�����ļ����Կ��ƾ��и��ߵ�Ҫ��, �Ա�֤�����ȷ�Ժ�������.
����Ŀǰ, ���������õ����������ݽ��л�����ע�͵����������ѷ�չ��һϵ���㷨��, ����С�������ȸߵ�������Ʒ���ݼ��ϵõ��˽Ϻõ�Ӧ��. ����ѧ�����������Ҳ������һ���ij���. �Ϻ�������Ϣ�����о�����л�غ��й���ѧԺ�Ϻ�������ѧ�о�Ժ����ѧ�Ŷ�[82,83]������ʹ��������������ݿ��С��ĸ߾����������ݽ����˳��Բ������� iGepros, �ɹ�Ӧ�õ�����͵����ʵ�����ע�͵��о���. ֵ��һ������й���ѧ�һ���������쵼���� HUPO ��֯�Ĺ���Ⱦɫ�嵰������ƻ�(chromosome-centric human proteome project, C-HPP),ּ��ͨ�����ʺ����Ը���Ⱦɫ���ϻ������ĵ����ʲ�����м���, ����һ�����ƻ������ע��. �ڸüƻ���, �й��Ŷ����й������ž�����ҽѧ��ѧԺ���������������о����ġ�������ѧ�������������о��������ϴ�ѧ����, ���ɺظ��������Mԭ����˹�桢����褵��˷ֱ����Ρ��������� 1, 8 �� 20 ��Ⱦɫ���ϱ������ĵ����ʲ���ļ�������, ���������Ŭ��, ȡ���˽��Խ�չ, �Ѽ��� 62%���ҵ�������������������, ����������ϵ���µ�©ע�ͻ���[79,84~86].
����(3) �����ʷ�������εļ������ʿ�.
���������ʷ��������(PTM)�ļ������������ѵ�: (��) ʵ������. PTM �ļ�������ǻ�ѧ����ˮƽ, �Ҵ���ʱ���, һ����˲ʱ���ֻ�̬�仯, ���������������ȵķ�����ѧ��⼼���ſ��ܲ���; (��) ������. �������������ͷḻ, ��ÿ�ֵ����ʵĶ��������л������ܷ�������, ʹ��PTM�ļ�������“��ϱ�ը”����,���ڴ���. ��������ݿ�����������Ԥ��ָ����������, �������������ĵ��Ķ�������������θ������ܵ�����, ֻ�ܼ������ݼ��еIJ�����������.
����Ȼ����ʹ����˫�߾���������(���� HCD), ͼ�Ľ�����Ҳֻ�� 50%����, ���, ������Ϊδ������ͼ�������̺��ŷḻ���µ��ס��ɱ���С�������ͻ��ͷ����������Ϣ, ؽ���ھ�. Ŀǰ�����ձ���Ϊ, δ�����Ķκ������Ķ�����Ʒ����ͬʱ���ڵ�,���ͨ���ھ��Ƶ���ֵ��Ķ�ĸ�����������ɫ�ױ���ʱ���, ���������ٷ���DZ�ڵ���������. ������һԭ��, �й���ѧԺ���㼼���о�������[87]�����������ھ� DeltAMT. ���������ö�ά��˹���ģ�ͽ��н�ģ, ������� D-score ��������������, ������Ч�ضԵ�һ���λ������ν��м���. ��ʵ��������, DeltAMT �Ը߷������(����һЩ��������)�нϺõļ���Ч��, ���Եͷ�����ε��ھ��Դ���һ������.
�����ڶԸ������ض��������͵�ʵ�����ݽ��д���ʱ, ��������μ����������������ƶԼ�����ȷ��Ҳ��Ϊ��Ҫ. ��ͬ���ữ�Ķξ��в�ͬ��������Ϊ. �����Ŷ�[88]�����������, �����ữ�������ײ����������˷�����˲���, �����ݶ�����ͼ�Ƿ������Զ�ʧ�彫���ữ�Ķν��з���, ÿ�������, ��������ữ�Ķμ����ķֱ��ʺ����ữ�����ʲ���ĸ��Ƕ�, ��������ữ������������, ʵ���˸�Ч�ļ���. ���Ŷӻ���չ������Ѫ���Ѽ��������������ݿ�, �����ö����ѿ��ʿصIJ�����������ữ�Ķεļ���������, Ҳ�������ѿ�ʱ��, �ٽ���ѪҺ���ữ��������ѧ���о�[89]. �������ữ��������Ĵ����ݼ�, ����ѧ����[90]�������ữλ���ڼ��������ض����ܷ��ӵı����Ը��������������Ӻ����������. ����, ��Ϊ���ữλ���ڼ�����Ľ��������з�������Ҫ������. Ҧѩ���Ѧ�����[91]�ֱ������켤ø�����ữλ����ϢԤ���(GPS2.0)�����˵��������̬��(single nucleotidepolymorphisms, SNP)����, ����Լ 70%������ SNPλ����DZ�ڵ����ữ SNP. ��Լ�� 74.6%��DZ�ڵ����ữ SNP �����˼�ø������ص����ữλ��ĸı�,������ֱ�Ӳ�������ȥ�����ữλ��. ��Щ��������������������༲�������еĻ������, �����õ�Ϊ���Ի�ҽ�Ʒ���.
����(4) �����������Ԥ��. ������������õ��о������ڵ�������������������̽��, ��ʾ�����ʵ�����ѧ����. ��Ŀǰ���������������Ԥ����о��ı���. �ڿ��������������Ԥ��� PRINCESS ֮��, �������������о�������Ŷ�[92]����̿����˵������������Ԥ��� SLIPPER. SLIPPERͨ�����Ϲ���ע�ͺ��������˽ṹ����, ��������ɸѡ�� Logistic �ع��������ý���Ԥ�ⷢ��, ������õ����������ں��и���Ľṹ��, ������Ҳ������, ����ѧ�����ϸ�������ø�ࡢ���һ����ҩ��е�ȵ�����. ������Щ������������������ж�ռ�ݹؼ��ڵ�, ��˵������������Ԥ���о���Ϊ��������������繹������֤��������.
����(5) ��������ѧ�о�������ݿ�����о���չ. ������ͨ���������ݵĴ����������������ݷ������������չ. �ڻ�������˼����Ͷ��������, ��Щ�������̺��ķḻ���﹦����Ϣ���ھ�ͬ����Ҫ������Ϣѧ��֧��. л���Ŷ�[93]ͨ���ռ����� 20 �����֢�� 331 ��ʵ������, �ṩ���˰�֢���쵰���������ݷ����ɹ��ο������ݿ�. ���Ѻ��Ŷ�[94]ͨ�������ھ���˹�ȷ��, �������˺Ͷ������Һ�����������־�����ݿ� UPB, ���ҷ��ֲ�ͬ���������������־��ĵ��ص��ʿ����벻ͬʵ���ҵ�ʵ�����̲����й�.
�������Ѧ���Ŷ�[95]�ڵ����ʵķ��ػ���������Ԥ�ⷽ�濪����һЩ�����ݿ�, �ٽ��˷�������ε�ע���о�. ����, ͨ�������ھ���˹�ȷ��, �ռ���26 �� E1, 105 �� E2, 1003 �� E3 �� 148 ��ȥ���ػ�ø��ȥ��ػ�ø����Ϣ, ��� E3 �ķ����о�, ���չ����˷��ؽ�Ϻ���ؽ�����ݿ�(ubiquitin andubiquitin-like conjugation database, UUCD). �����ݿ���¼�˺�� 70 ������������ֵ�Լ 6 ����ø����Ϣ.
�����������, ��������CPLA��������������λ�����ݿ�[96,97], �Լ���¼�˰��������ڵ� 7 ��ģʽ�������ǧ����ϸ���ֻ�����˿���ѹ��������м��塢���������˿�����ϵĵ��������ݿ� MiCroKit, ΪȾɫ����صĵ�������ѧ���о�����������.
����3 ��չ��
�������й���������ѧ�Ҳ�иŬ�����ֿ�ѧ����Ķ���֧����, �й��ĵ�������ѧ�о��ڹ�ȥ�� 3 ���м����������������١����չ��̬��. �й���������ѧ�о��Ŷӳе��Ĺ��ʸ��൰������ƻ�ȡ���˽��Գɹ�, �����˹������ٵ�������ѧ�ķ�չ,��Ӱ���ź����Ĺ������൰������ѧ�о�. �ڹ��ʸ��൰������ƻ�ʵʩ�������γɵ�˼·�����ԡ�������ϵ���˲Ŷ���Ϊ�ո��������й����൰������ƻ��Ŀ�չ�춨�˼�ʵ�Ļ���. ����Ԥ��, ͨ��δ�������Ŭ��, ���൰������ѧ���о��ɹ��ڲ��������벡�����̵ķ��ӻ��Ƶ��о��н��ᷢ�Ӹ��ӻ���������, ��Ϊ����Ľ�����ҵ��������ѧ�ķ�չ�춨����.
������л
�������ĸ�л���������͵ط������Ƽ�����, �ر��ǹ��Ҵ��ѧ��ʩ�ƻ���������Ȼ��ѧ�����Լ������������Ե�������ѧ��չ��֧�ֺ�����. ͬʱ��л CNHUPO ��֯�Լ��й���������ѧ�ҵ�֧�������, �ر��������е������ڵ�������ѧ��չ��������Ŭ���빱��. ����ƪ������, ���� CNHUPO ��Ա�г�Ч�Ĺ���û�б��ἰ, �ڴ�һ����ʾ�����ǵľ������л.
�����ο����ף�
����1 Gao X, Zhang X, Zheng J, et al. Proteomics in china: ready for prime time. Sci China Life Sci, 2010, 53: 22–33
����2 He F. At a glance: proteomics in china. Sci China Life Sci, 2011, 54: 1–2
����3 �ظ���. ����ʱ����“������ѧ”(����). �й���ѧ: ������ѧ, 2013, 43: 1–15





