摘 要: 为更准确识别核小体定位,本文提出一种基于Z曲线理论(Z-Curve)的卷积神经网络(CNN)方法,称为ZCN方法。ZCN方法以Z曲线三维坐标矩阵表示核小体序列特征,通过十倍交叉验证,进行卷积神经网络方法进行模型训练和验证,使用标准评估指标进行性能评价。结果表明:ZCN方法在酵母中具有良好的识别效能,敏感性Sn、准确性Sp、ROC曲线面积分别为92.4%、90.2%和0.970 4,可推广到人类、线虫和果蝇的核小体定位识别中,其ROC曲线面积分别为0.796、0.940和0.772,与其他方法比较,进一步证实ZCN方法具有较好的识别效能和可推广性。在酵母全基因组进行核小体定位预测,发现16条染色体的预测准确率均值为78.83%,在基因GAL和GAL10中进行核小体定位预测,研究了降低假阳性的方法,给出了预测核小体定位的图谱。ZCN方法为研究核小体定位识别、预测及功能分析提供了有价值的方法和指导。
关键词 : 计算生物学;卷积神经网络; Z曲线理论;核小体; DNA序列;连接区;
Abstract: In order to improve the recognition accuracy of nucleosome positioning, we applied a method, that convolution neural network method based on the Z-curve theory was used to identify nucleosome DNA sequence and called ZCN method, the z-curve three-dimensional coordinate matrix was used to represent the features of nucleosomes, the convolutional neural network was trained and verified by nucleosome sequences and linker sequences through 10-fold cross validation, and use standard evaluation indicators for performance evaluation.The results show that the convolutional neural network method has a good recognition performance in Saccharomyces cerevisiae, and Sensitivity Sn, accuracy Sp and ROC curve areas were 92.4%, 90.2% and 0.9704, respectively. It can be applied to the localization of nucleosomes in H.sapiens, C.elegans and D.melanogaster, the ROC curve areas were 0.796, 0.940 and 0.772, respectively, and compared with the other four methods, it is further confirmed that ZCN method has better identification efficiency and generalization. In the whole genome of S. cerevisiae, the prediction accuracy of nucleosome positioning was found to be 78.83% for the mean of 16 chromosomes. ZCN was used to predict nucleosome positioning on GALl and GAL10 genes, which the method of reducing false positive was studied and the map of predicting nucleosome was displayed. In conclusion, ZCN method provides valuable methods and guidance for the researching nucleosome positioning, recognition, prediction and function analysis.
Keyword: computational biology; convolutional neural network; Z-curve theory; nucleosome; DNA sequence; linker;
卷积神经网络(convolutional neural networks, CNN)已被广泛地应用于很多研究领域[1],如图像分类、人脸识别、交通标志识别等[2]。本文将卷积神经网络用于生物数据核小体DNA序列的识别。核小体是真核生物的染色质基本结构单元[3],由约147 bp的DNA双链缠绕组蛋白八聚体约1.75圈形成,是染色体的一级结构[4]。相邻核小体通过称短DNA序列连接,其范围为10 bp~100 bp。DNA序列特征一直被认为是核小体定位的重要因素。核小体参与染色质形成[5]、拮抗转录因子[6]、抑制基因表达[7]等重要的生物学过程,其DNA序列的精确定位不仅影响基因表达调控[8],对DNA复制[9]、DNA修复[10]及重组[11]等也有重要作用。当前国内外核小体定位研究大多针对开发分类算法,例如通过信息熵[12]、碱基对偏转角度[13]等方法来表示核小体DNA序列特征[14],进而训练分类器。本文基于Z曲线理论将核小体DNA序列转换为三维空间曲线坐标,应用卷积神经网络模型训练和检验,获得了较好的分类效能,为今后的核小体定位研究提供一些参考。

1 、基于Z曲线理论的卷积神经网络方法(ZCN)
1.1 、Z曲线理论及转换公式
Z曲线理论基于正面体表示碱基对称性[15],将DNA 序列用4 种字母符号表示为一维序列,利用这种形式来表示任意长度的DNA 序列,能够显示出DNA序列的新特征,且根据正四面体的对称性可以证明,每条序列对应唯一一条Z曲线。对于一条长为N的单链DNA序列,它的三维坐标可表示方法为:
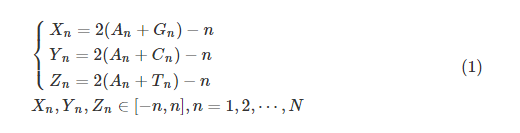
每次从第1个碱基到第n个碱基,分别统计1~n这个子序列中4种碱基积累出现的次数,记为An、Cn、Gn、Tn。Xn、Yn、Zn的取值范围为[-n,n],对其进行标准化处理, 将Xn、Yn、Zn的值都除以n得到xn、yn、zn,使其范围处于[-1,1]:

1.2、 卷积神经网络分类器
卷积神经网络是一种前馈神经网络[16],包括卷积层(convolutional layer)和池化层(pooling layer),布局更接近于真实生物神经网络,能降低特征提取和分类过程中数据的复杂程度[17]。卷积神经网络的构建包括创建卷积层、创建线性整流层、创建池化层以及创建全连接层4个步骤。
本文基于Z曲线理论,应用卷积神经网络提出一种新的核小体定位识别方法,简称为ZCN,该方法的流程图如图1所示。分类器的构建过程使用R软件包“mxnet”进行训练和验证,采用十倍交叉验证方法进行效果评估,取10次验证的平均结果为一次最后结果,同时,为了减少由于随机分类而带来的结果误差,随机重复进行50次十倍交叉验证。具体过程如下:将Z曲线模型投入卷积层构建模型,卷积核大小为3,并选定卷积核个数为300创建卷积层;然后加入非线性函数即双曲正切函数创建线性整流层;再采用最大池化,步长设置为1创建池化层;最后,每一个结点都与上一层的所有结点相连,用来把前边提取到的特征综合起来,最终得到全连接层,并用“softmax”函数构建分类器模型,使用验证集数据进行分类器检验;最后,通过验证数据集进行验证。
图1 基于Z曲线的卷积神经网络流程
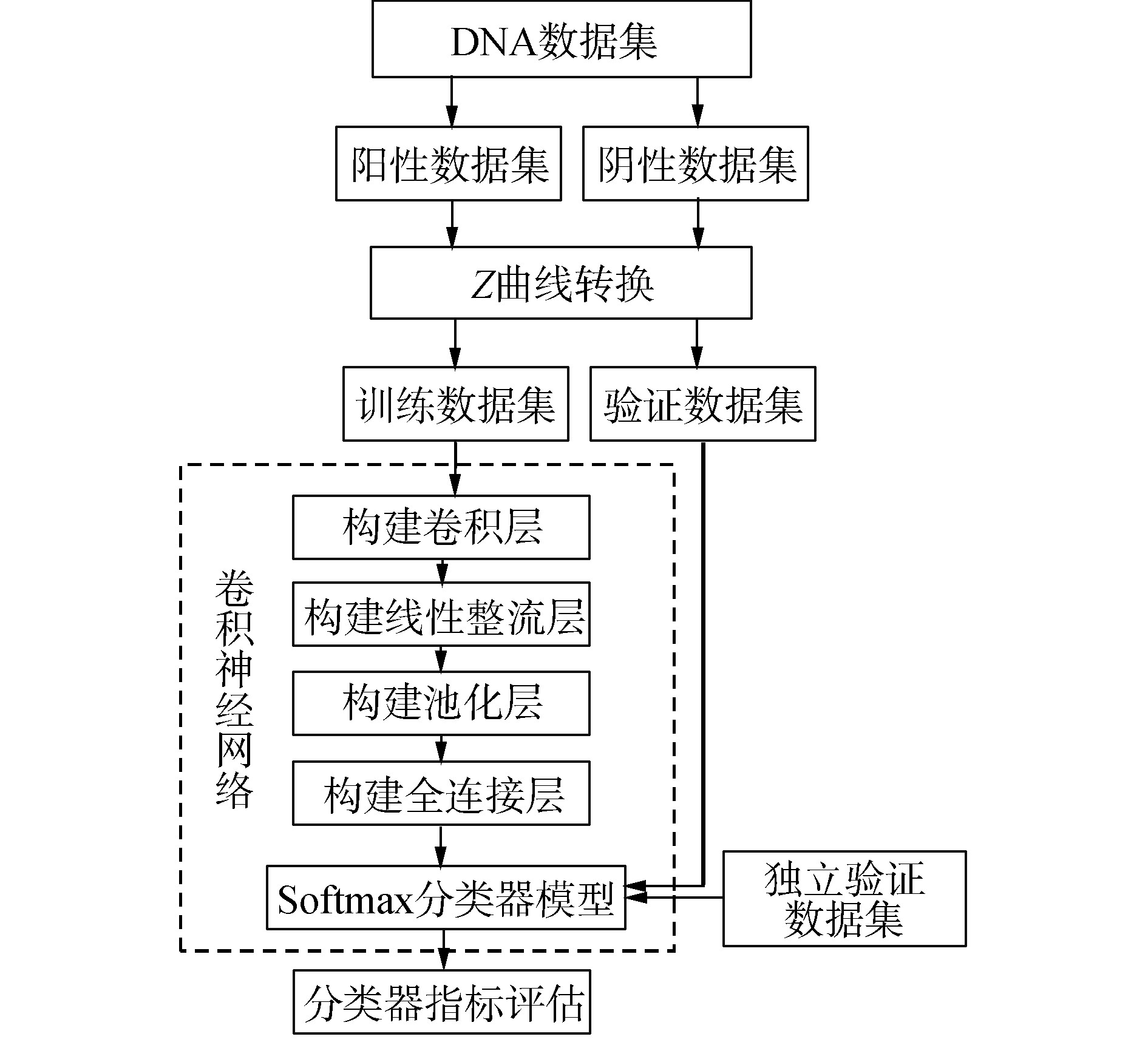
Fig.1 Flow chart of convolutional neural network based on Z-curve
1.3、 分类器模型性能评估
为评价ZCN方法的分类效果,使用敏感性(sensitivity, Sn)、特异性(specificity, Sp)、准确率(accuracy, Acc)和Matthews相关系数(matthews correlation coefficient, MCC)及ROC(receiver operating characteristic)曲线面积AUC(area under ROC)来作为评价参数[18],前3个指标通常被用于在统计预测理论中从不同角度衡量预测系统性能为:

式中:TP表示真阳性(true positive, TP)数量;FP表示假阳性(false positive, FP)数量;TN表示真阴性 (true negative, TN) 数量;FN表示假阴性(false negative, FN)数量。
2、 实验结果
2.1、 数据来源及预处理
2.1.1、 酵母数据集
从文献[19]中获得酵母(Saccharomyces cerevisiae, S.cerevisiae)核小体数据,含有5 000条核小体DNA序列作为阳性数据集,与5 000条连接DNA序列作为阴性集,每条序列长为150 bp, 记为数据集S1;同时,采用文献[20]中的1 880条核小体DNA序列集和1 740条连接区DNA序列集,记为数据集S2。
2.1.2、 人类、线虫和果蝇数据集
从Guo文献中获得人类(H.sapiens)、线虫(C. elegans)和果蝇(D. melanogaster)的数据集[21],人类共有2 273条核小体DNA序列集和2 300条连接区DNA序列集,线虫共有2 067条核小体DNA序列集和2 108条连接区DNA序列集,果蝇共有2 400条核小体DNA序列集和2 350条连接区DNA序列集,用于检验ZCN方法分类效能和可推广性。
2.1.3、 酵母全基因组数据
从UCSC数据库获取酿酒酵母全基因组序列数据[22],包含17条染色体序列,版本:SacCer_Apr2011 sacCer3),使用其中16条染色体数据;另外从SGD(Saccharomyces Genome Database)数据库获得酵母基因GAL1和基因GAL10的DNA序列数据,用于核小体预测。
2.1.4 、酵母全基因组核小体数据集
从Xu Zhou与Erin O′Shea的文献中获得61 532条酵母核小体位置信息数据[23],这是一套全基因组的核小体数据集,用于检验预测结果。
2.2、 分类器识别结果与分析
2.2.1 ZCN方法在酵母数据集中的实验结果
酵母数据集S1的核小体DNA序列和连接区DNA序列经过卷积神经网络进行训练和验证,结果如图2(a),敏感性Sn、特异性Sp、准确率Acc和MCC值分别为0.91、0.88、0.90、0.80,ROC曲线下面积AUC值为0.96,面积最高值达到0.970 4,如图2(b),这表明ZCN方法在酵母的核小体定位识别中的效能良好,能够较好地识别出核小体序列与连接区序列。
图2 ZCN方法实验结果和ROC曲线(S1)
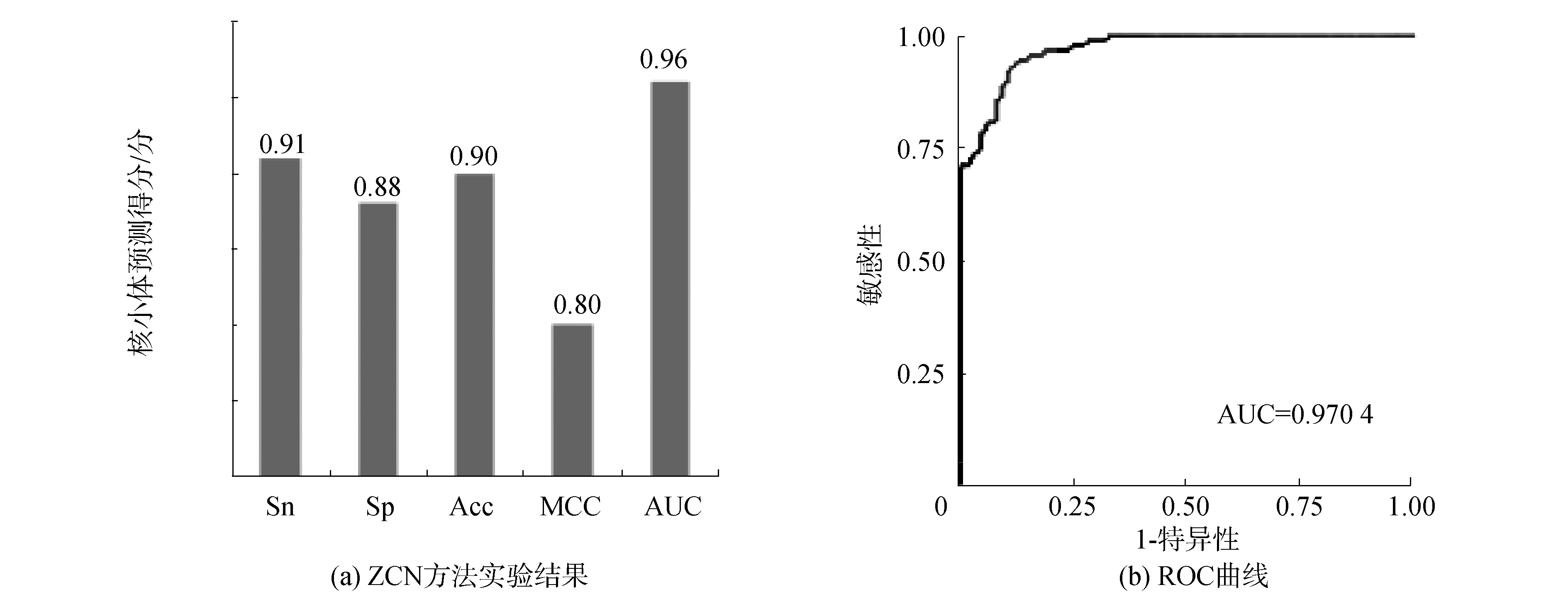
Fig.2 Results of ZCN method and receiver operating characteristic(S1)
酵母数据集S2的核小体DNA序列和连接区DNA序列,分别经过Z曲线转换得到三维空间坐标矩阵数据集,投入训练出的分类器中进行分类,通过十倍交叉验证,结果如图3(a)所示,Sn、Sp、Acc和MCC分别达到0.97、0.84、0.90、0.82。ROC曲线面积AUC值最高达到0.972 3,如图3(b)所示,表明ZCN方法在酵母核小体定位中再次取得较好识别效果,且各项性能指标稳定。实验表明ZCN方法在酵母中具有较好的应用效果。
图3 ZCN方法实验结果和ROC曲线(S2)
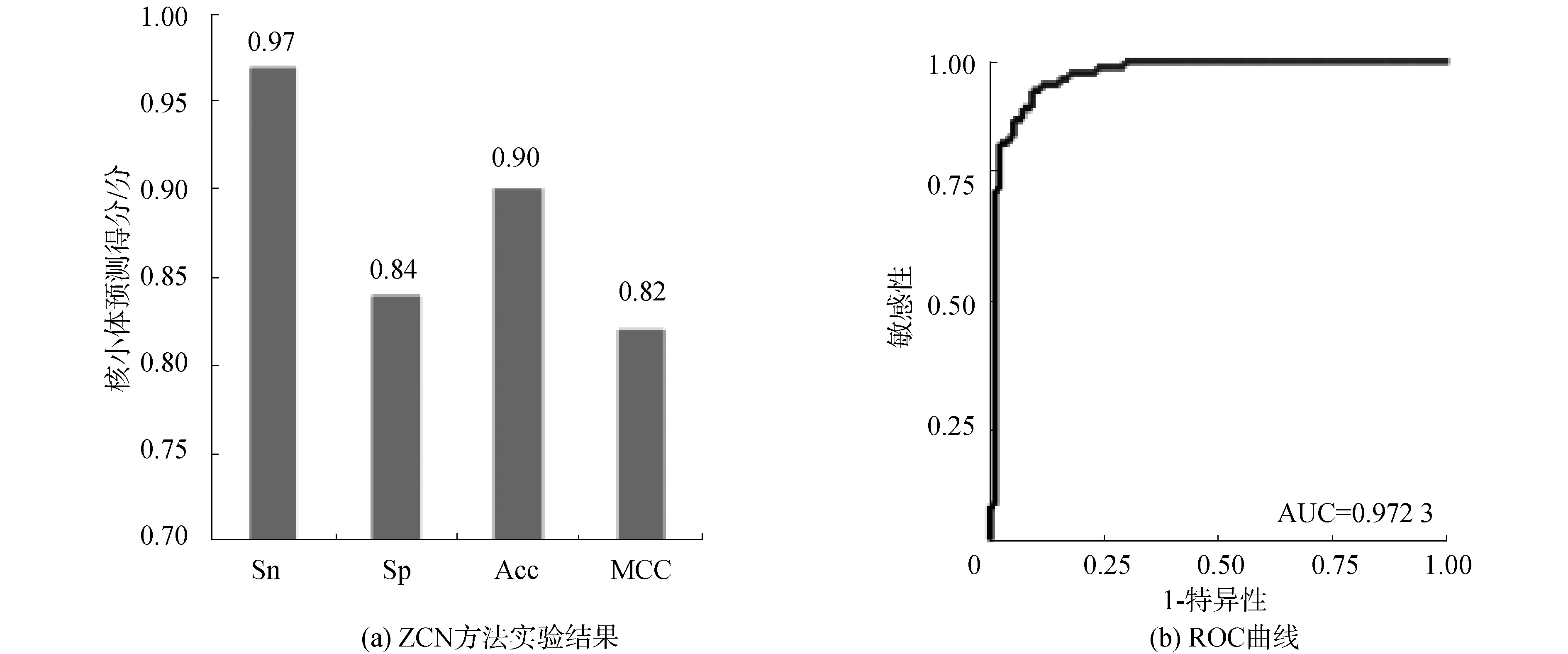
Fig.3 Results of ZCN method and receiver operating characteristic(S2)
2.2.2、 ZCN方法在人类、线虫和果蝇中的实验结果
应用ZCN方法识别人类、线虫和果蝇3个物种的核小体定位,得到人类、线虫和果蝇的ROC曲线下面积AUC值分别为0.796、0.940和0.772,如图4所示,Sn、Sp和Acc值如表1所示。ZCN方法不仅可应用于酵母也可应用于其他多个物种的核小体定位识别,分类效能良好且具有可靠的推广性。
图4 人类、线虫和果蝇的ROC曲线面积
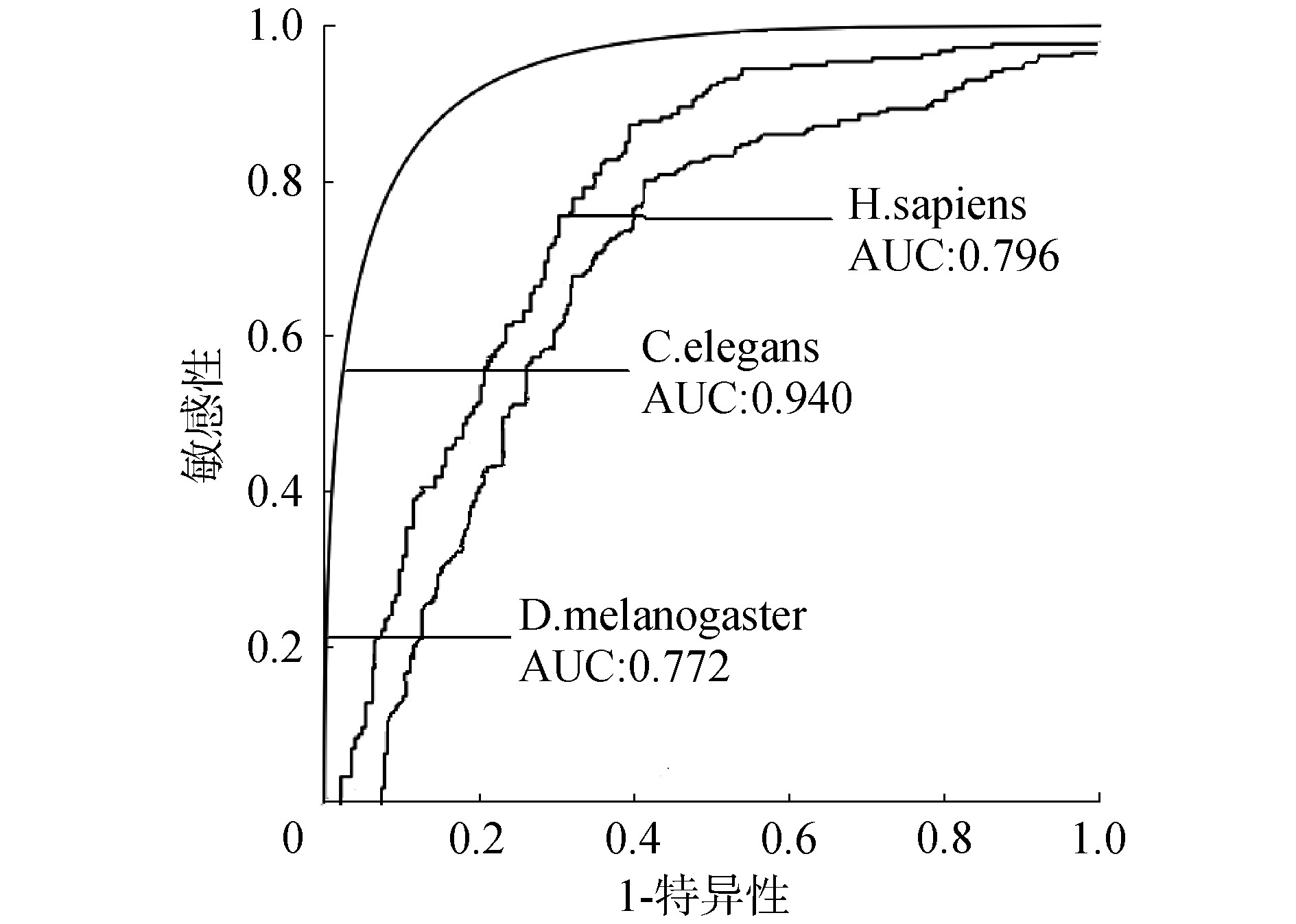
Fig.4 ROC curve areas for H.sapiens, C.elegans and D.melanogaster
2.2.3、 ZCN方法与其他识别方法的比较
将ZCN方法与其他方法进行结果比较,包括iNuc-STNC方法[21]、iNuc-PseKNC方法[18]、3LS方法和LeNup[14],这4种方法没有酵母实验结果,因此只进行人类、线虫和果蝇的核小体定位识别结果比较;另外,LeNup方法采用20倍交叉验证,而3LS、iNuc-PseKNC和iNuc-STNC方法采用Jackknife检验方法,ZCN方法采用10倍交叉验证方法,其训练数据集在样本大小和训练次数上均小于上述4种方法,如表1所示。
表1 ZCN方法与其他方法的实验结果比较
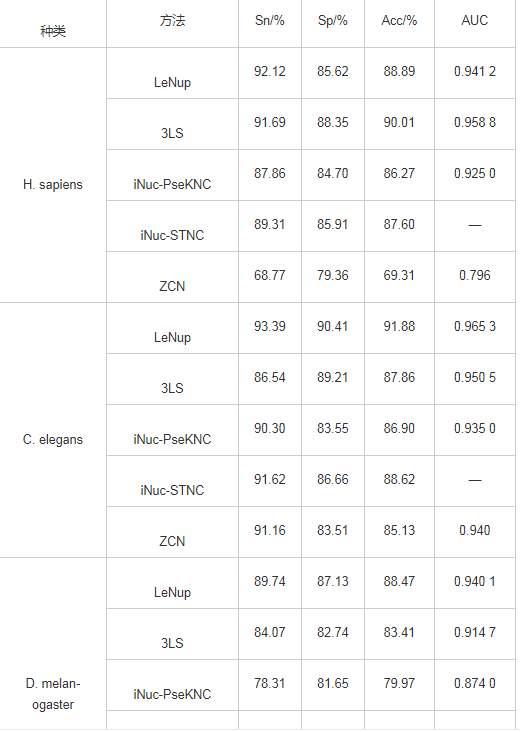
ZCN方法在人类和果蝇数据集中,敏感性Sn、特异性Sp、准确性Acc 3项指标略低,ROC曲线面积AUC值分别达到0.796和0.772,而iNuc-STNC方法没有给出AUC值;在线虫数据中,ZCN方法的敏感性Sn高于3LS方法,特异性Sp、准确性Acc与iNuc-PseKNC方法基本一致,ROC曲线面积AUC值0.940略高于iNuc-PseKNC方法。ZCN方法在训练数据集大小和训练次数均小于其他4种方法,但在单项指标上表现较好,且各项指标稳定,特别地在酵母中取得较好实验结果,表明ZCN方法有识别较好效能和可推广性。
2.3 、酵母核小体预测结果与分析
2.3.1、 核小体序列预测候选序列集
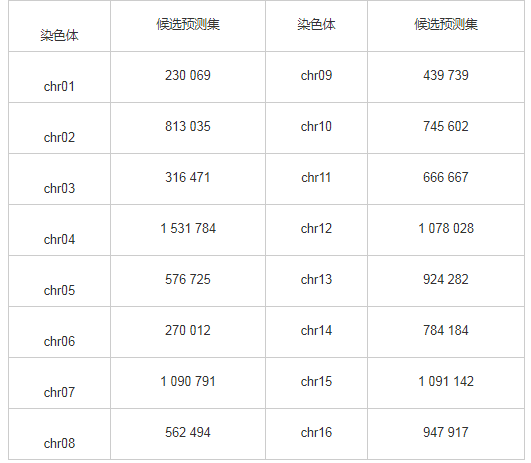
通过滑窗法,设置滑动窗口大小为150 bp, 步长为1 bp, 按染色体提取出核小体DNA序列候选预测集,如表2所示,共得12 068 942条DNA序列作为候选预测集,将每条DNA进行Z曲线转换得到坐标矩阵,投入到ZCN方法训练的模型中进行预测。
表2 酵母全基因组核小体定位候选预测集
2.3.2 、核小体预测去冗余筛选方法
滑窗法获得候选序列集存在大量相似序列和冗余数据,为减少预测结果中的重复和冗余,提出基于染色体上每个碱基位置的去冗余策略,对序列阳性集进行筛选,保留预测为核小体的DNA片段,将这些片段回拼至全基因组对应位置上。
筛选方法思想如下:1)每条染色体上的每条候选预测序列,除去首尾2条候选预测序列的75 bp碱基外,每条候选预测序列的每个碱基,将其扩展前后共150 bp序列提取出来投入ZCN训练模型中进行预测。若预测为核小体,则将这一碱基标记为核小体。因此,除了每条染色体序列最开始的149个碱基与末尾149个碱基,相当于在考虑周围序列信息的条件下,对染色体上单个碱基进行150次记分。碱基所对应的得分越高,该碱基前后共150 bp碱基所对应的150条序列被分类为核小体序列的数量越多,即这个位置的碱基更倾向于落到核小体序列片段;2)如果一条DNA片段被分类为核小体,则该序列对应碱基150 bp范围内的所有碱基得分加1,否则加0,在对所有序列进行分类之后,所有位置的碱基的得分范围为0~150,设定初始阈值75,当每个位置的碱基得分阈值大于或等于75时,认为该位置碱基更加倾向于落在核小体区域,逐渐提高阈值,找到可以降低假阳性的更加严格的阈值,最后统计筛选出得到去除有重复的相邻候选序列,得到预测的核小体数量,如图5所示,经过28次计算,最后选择阈值为93筛选所得9 229 869个碱基位置作为核小体预测结果。
2.3.3、 核小体预测结果验证
为检验其与验证集在单条染色体上的核小体结果的一致性,使用一套独立数据集(61 532条核小体序列)来验证结果,绘制酵母单条染色体上预测碱基与实际碱基位置统计图。如图6所示,发现不仅每一条染色体上的2个数据量大小相近,全基因组上的整体数据趋势也相同,如图6(a)所示;为了精确观察具体差异程度,计算出每条染色体2类数据的差值在真实碱基数量上的比例,结果显示差值比例都小于4%,最大值为3.7%,最小值仅为0.1%,如图6(b)所示,预测结果与验证数据有较好的一致性,表明核小体去冗余筛选方法得到的阈值具有可靠性。
图5 候选预测结果阈值筛选
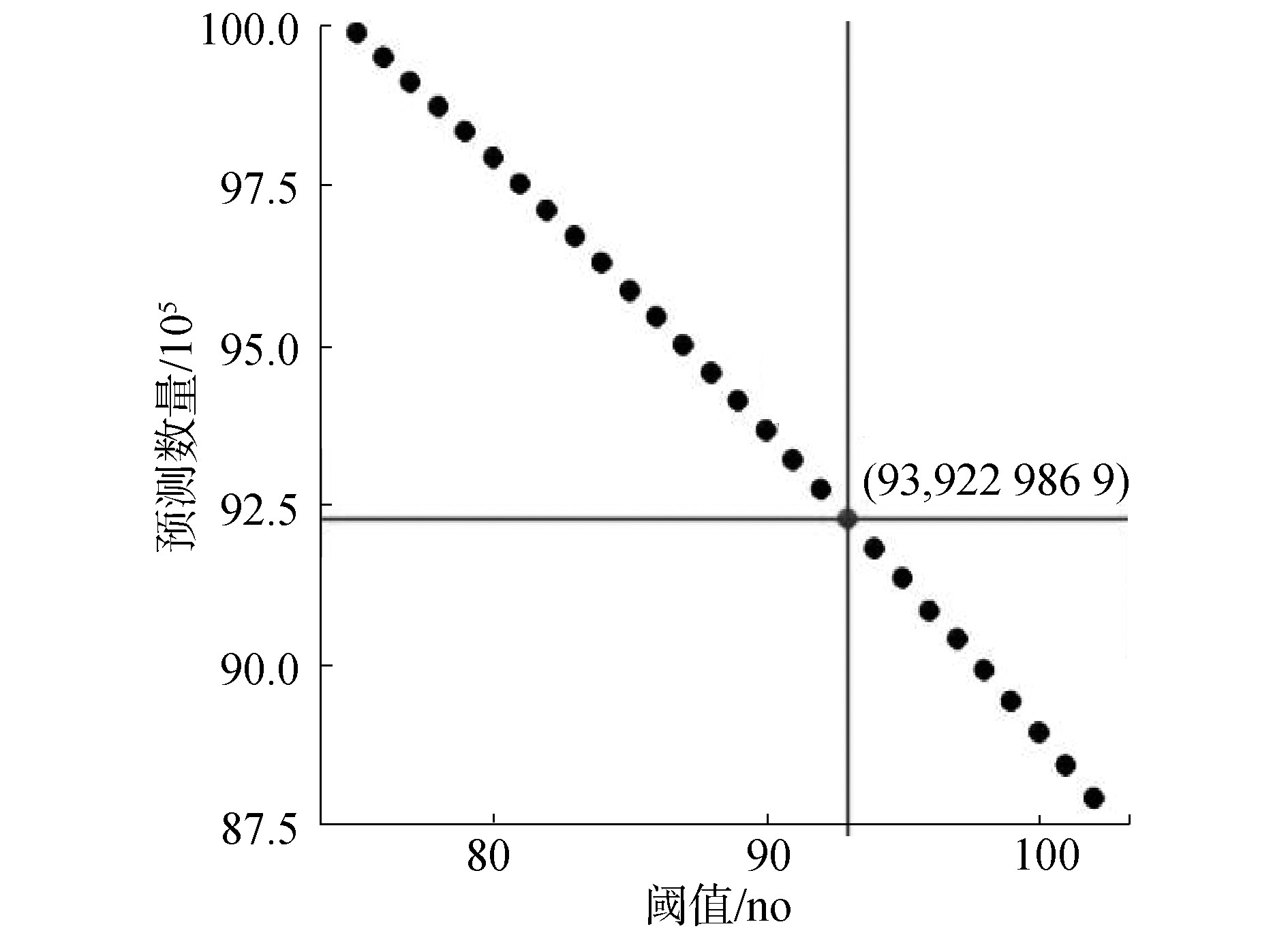
Fig.5 Threshold selection of candidate prediction result
图6 预测位置与实际位置的验证统计
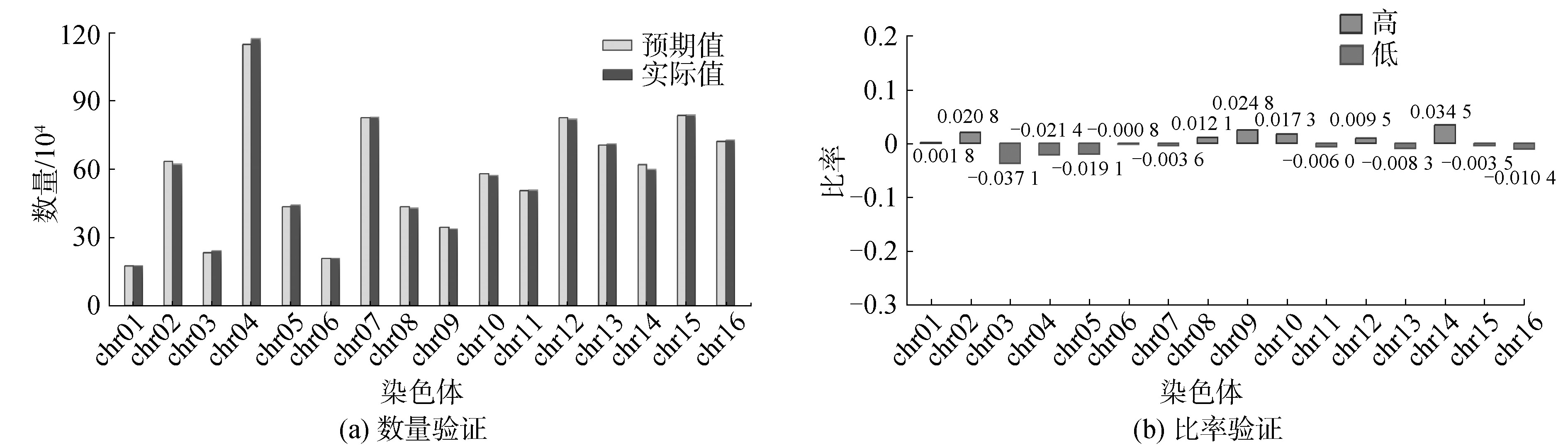
Fig.6 Statistics of predicted position and actual position
计算单条染色体预测位置与实际位置的交集,以检验核小体定位的准确性,可见位置的重叠程度较高,如表3所示。
表3 酵母核小体定位的全基因组位点验证
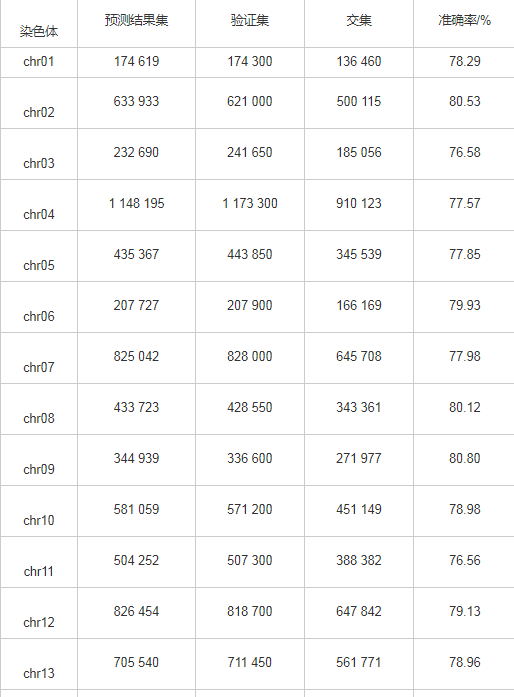
单条染色体交叠碱基数量真实验证数据集碱基数的比例最低为76.56%,最高81.49%,平均值为78.83%,根据比例值,对预测结果集与验证集做Wilcoxon秩和检验,P-value为 0.690 5,差异不显着;根据位置,对预测结果集数据与验证集的核小体起始和终止位置,计算其皮尔森相关系数(Pearson correlation coefficient)以验证预测结果与验证结果的相似性,发现每条染色体上的皮尔森相关系数值均大于0.99,P-value<2.2×10-16,这充分说明预测结果集与验证集的相关性接近于100%。根据结果分析,发现核小体序列能够很大范围地被预测出来,通过阈值筛选后,其验证结果较好,说明ZCN方法可以进行全基因组核小体定位预测,并且在训练集大小为5 000条核小体序列和5 000条连接序列的情况,不仅完成全基因组6万多条核小体序列预测,并取得78.83%的预测准确率,结果证实ZCN方法预测效果较好。
2.3.4 、酵母基因GAL1与基因GAL10的预测结果
从SGD数据库获得基因GAL1和GAL10的DNA序列数据及位置数据,用ZCN方法在这两个基因上进行核小体定位预测和验证,预测过程与全基因组上核小体预测过程一致,GAL1原基因长度为1 587 bp(chrII:279021-280607),加上下游1 kb, 共为3 587 bp(chrII:278021-281607),GAL10原基因长度为2 100 bp(chrII:276253-278352),加上下游1 kb, 共为4 100 bp(chrII: 275253-279352)。
预测结果如图7所示,计算该基因上预测位置与实际位置的交集,及交集在预测集与真实集中的占比。
图7 预测位置与实际位置的验证统计
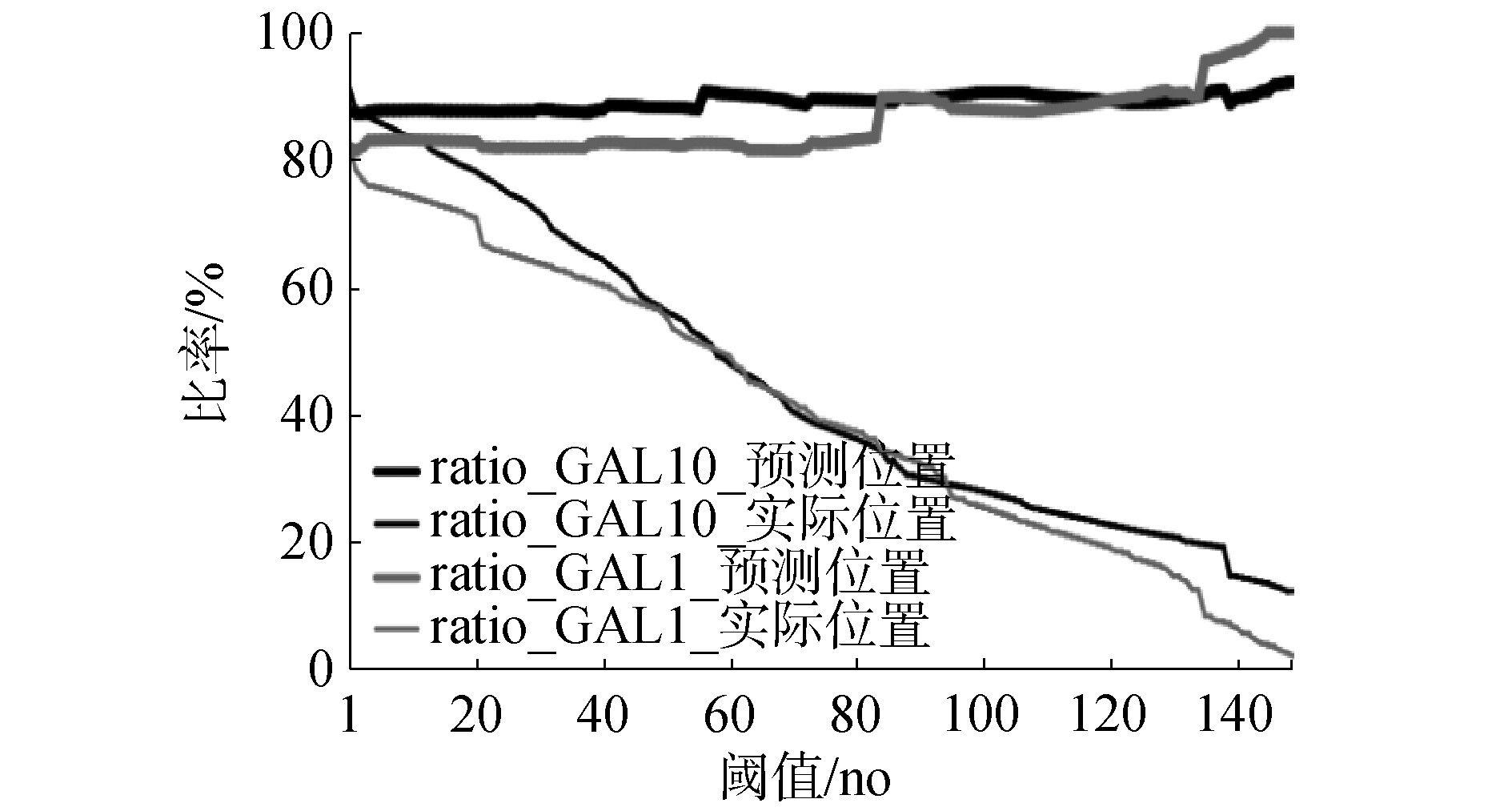
Fig.7 Statistics of predicted position and actual position
图7中,粗线表示预测核小体碱基中交集位点的含量,即预测核小体碱基的准确率;细线表示在真实核小体碱基中交集位点的比例,也可以表示每个阈值下核小体的检出率,它越来越低是因为随着阈值升高,预测碱基位点数量逐渐降低,导致交集数量减少,但作为分母的真实核小体碱基数量一直不变。当阈值设为1时,表示以卷积神经网络模型一次分类即为预测结果,此时在基因GAL10序列中有3 374个碱基被预测为核小体位点,其中有2954(约87.55%)个为真实的核小体碱基位点,占真实的核小体碱基位点数据集(3 241个)的91.14%。而在基因GAL1序列中有2 699个碱基被预测为核小体位点,其中有2 213(约81.99%)个为真实的核小体碱基位点,占真实的核小体碱基位点数据集(2 700个)的81.96%。可见位置的重叠程度较高,说明在不考虑假阳性的情况下,核小体序列能够很大范围地被预测出来。
为了降低假阳性,逐渐提升阈值来综合考虑多次分类的结果,虽然核小体的预测位点数量与碱基交集数量逐渐减少,但该阈值下预测集中的准确性却逐渐增高至92.53%,甚至100%(GAL10中:准确性最低为87.25%,最高为92.53%,平均值为89.16%;GAL1中:准确性最低为81.38%,最高为100.00%,平均值为86.25%),结果证实ZCN方法预测效果较好。尽管真实的核小体位点数量的检出率显着降低,但是预测集中的准确性却波动相对较小,这意味着预测结果中假阳性比例不大,且今后也许可以根据核小体所需数量来确定阈值,即使将阈值设置为个位数,预测的准确性都能高于81%。根据预测核小体碱基位点的得分,绘制峰值图谱,如图8所示,结果显示在基因GAL1周围上下游出现7个峰值,基因GAL10周围上下游出现11个峰值,即预测的核小体定位数量,这与理论分析基本一致,再次说明ZCN方法的预测效能较好。
图8 基因GAL1和GAL10上的核小体定预测图谱
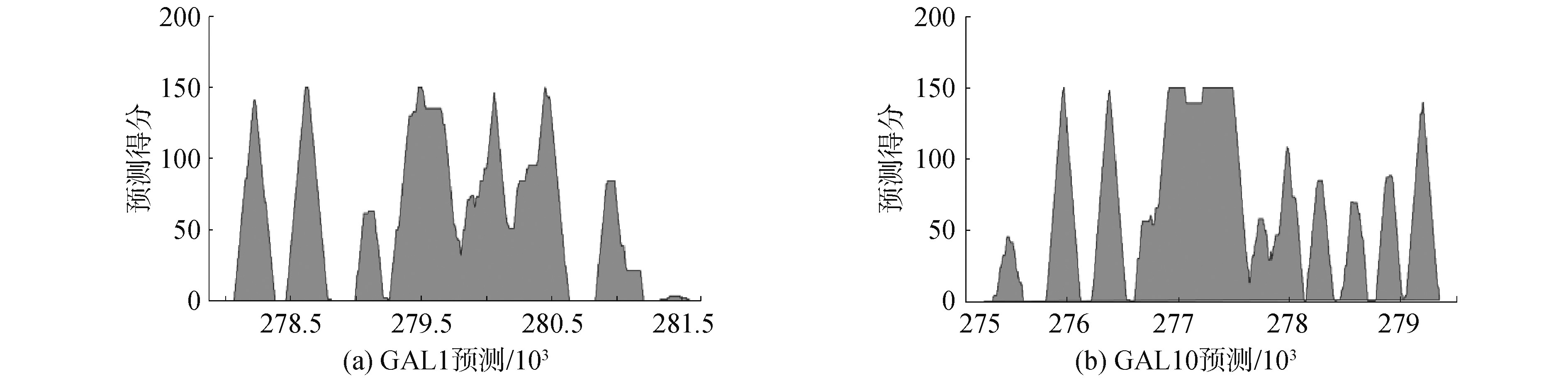
Fig.8 Predicted map of nucleosome positioning on GAL1 and GAL10 genes
3 、结论
1)将ZCN方法用于酵母核小体定位识别,通过2套数据实验,结果显示ZCN方法在酵母中取得较好的识别效果。
2)将ZCN方法能够推广到其他物种,包括人类、线虫和果蝇的核小体定位识别中,与其他4种方法进行性能比较,结果显示ZCN方法能够很好进行物种推广。
3)将ZCN方法用于酵母全基因组核小体定位预测,又提出一个基于碱基位置的阈值筛选方法,既获得高质量的核小体偏好碱基,又降低数据冗余性,将筛选后的预测结果与实验获得的核小体数据(验证集)比较验证,同时通过预测基因GAL1和GAL10周围的核小体位点,进而获得核小体定位的位置分布情况,均显示出预测结果集与验证集的具有较好的一致性和较高的准确,说明ZCN方法能够很好地完成核小体定位预测。
ZCN方法获得较好的实验结果,是由于Z曲线的三维坐标矩阵很好地展现出DNA序列特征,卷积神经网络很好地完成了这些特征的训练,因此,实验结果显示各项性能指标都取得不错效果。ZCN方法对核小体定位预测和核小体功能研究具有重要的参考和指导意义,特别地,对于深入理解基因表达的后续步骤以揭示控制核小体定位所涉及的机制也有重要作用。
参考文献
[1] SARIGOL M,OZYIL DIRIM B M,AVCI M. Diferential convolutional neural network[J] Neural networks,2019.116.279-287.
[2]张功国,吴建,易亿,等基于集成卷积神经网络的交通标志识别[J]重庆邮电大学学报(自然科学版),2019.31(04):571-577.ZHANG Gongguo,WU Jian,YI Yi,et al.Tafic sign recognition based on ensemble convolutional neural network[]. Journal of Chongqing University of Posts and Telecommunications(natural science edition),2019,31(4)-:571-577.
[3] TABERLAY P C,STATHAM A L,KELLY T K,et al.Reconfiguration of nucleosome- depleted regions at distal regulatory elements accompanies DNA methylation of enhancers andinsulators in cancer[J].Genome research,2014,24(9):1421-1432.
[4] FARMAN F U,IQBAL M.AZAM M.et al.Nucleosomes positioning around transcriptional start site of tumor suppressor (Rbl2/p130) gene in breast cancer[J]. Molecular biology reports ,2018,45<2):185-194.
[5] BUCKWALTER J M.NOROUZI D,HARUTYUNYAN A,et al. Regulation of chromatin folding by conformational variations of nucleosome linker DNA[J]. Nucleic acids research,2017 ,45(16):9372-9387.
[6] MURUGAN R.Theory of site-specific DNA-protein interactions in the presence of nucleosome roadblocks[J] Biophysical journal,2018, 1411):2516-2529.
[7] NOCETTI N,WHITEHOUSE I.Nucleosome repositioning underlies dynamic gene expression[J].Genes & development,.2016.30(6):660-672.
[8] BAI Lu,MOROZOV A V.Gene regulation by nucleosome positioning[J]. Trends in genetics,2010,26(11):476-483.
[9] EATON M L,GALANI K,KANG S,et al.Conserved nucleosome positioning defines replication origins[J]. Genes & development,2010,24(8):748-753.
[10] YING Hua, EPPS J,WILLIAMS R,et al.Evidence that localized variation in primate sequence pergence arises from an infuence of nucleosome placement on DNA Repair[J].Molecular biology and evolution,2010,27(3):637-649.
[11] BEVINGTON S,BOYES J.Transcription-coupled eviction of histones H2AH2B governs V(D)J recombination[J].The EMBO jourmal,2013,32(10): 1381-1392.
[12] XING Yongqiang,LIU Guoqing,ZHAO Xiujuan,et al.An analysis and prediction of nucleosome positioning based on information content[J] Chromosome research,2013,21(1):63-74.
[13] LIELEG C,KRIETENSTEIN N.WALKER M,et al. Nucleosome positioning in yeasts :methods,maps. and mechanisms[J] Bioinformatics,2015,124(2)-131-151.
[14] ZHANG Juhua,PENG Wenbo,WANG Lei.LeNup:learning nucleosome positioning from DNA sequences with improved convolutional neural networks[J] Bioinformatics,2018,34(10):1705-1712.
[15]张任,张春霆Z曲线,显示和分析DNA序列的直观工具[J]自然杂志, 1995,17(1):34-37 .ZHANG Ren,ZHANG Chunting. Z-curve: an intuitive tool for visualizing and analyzing the DNA sequences[J.Chinese journal of nature, 1995,17(1):34-37.
[16]刘超,张晓晖,胡清平图像超分辨率卷积神经网络加速算法[J]国防科技大学学报, 2019.41(2):91-97 .LIU Chao ZHANG Xiaohui,HU Qingping. Image super resolution convolutionneural network acceleration algorithm[J].Journal of National University of Defense Technology,.2019.41(2):91-97.
[17]杨军,王亦民基于深度卷积神经网络的三维模型识别[J]重庆邮电大学学报(自然科学版).2019,31(2)-253-260. YANG Jun,WANG Yimin. 3D model recognition based on depth convolution neural network[J] Journal of Chongqing University of Posts and Telecommunications(natural science edition),2019,31(2):253-260.
[18] GUO Shouhui,DENG Enze,XU Liqin,et al. iNuc-PseKNC:a sequence- based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition[J]. Bioinformatics ,2014,3011):1522-1529.
[19] CHEN Wei,LIN Hao,FENG Pengmian,et aliNuc-PhysChem:a sequence-based predictor for identifying nucleosomes via physicochemical properties[J].PLoS one,2012,7(10):e47843.
[20] CHEN Wei,FENG Pengmian,DING Hui,et al.Using deformation energy to analyze nucleosome positioning in genomes[J]. Genomics ,2016, 107(2/3):69-75.
[21]TAHIR M,HAYAT M.iNuc-STNC:a sequence-based predictor for identification of nucleosome positioning in genomes by extending the concept of SAAC and Chou's PseAAC[J].Molecular biosystems,2016, 12(8):2587-2593.
[22] KAROLCHIK D,BAERTSCH R,DIEKHANS M,et al.The UCSC genome browser database[J] Nucleic acids research,2003,31(1):51-54.
[23] ZHOU Xu,BL OCKER A W.AIROLDI E M,et al.A computational approach to map nucleosome positions and altermative chromatin states with base pair resolution[J.eLife. 2016,5:e16970.本文引用格式:崔颖,施丹丹, 徐泽龙,等卷积神经网络在核小体定位识别中的应用[J]哈尔滨工程大学学报, 2021,42(5):751-758 CUI Ying.SHI Dandan,XU Zelong,et al.Application of convolutional neural network based on Z-Curve theory in identifying nucleosome Positioning[J]. Journal of Harbin Engineering University,2021,42(5):751-758.





