摘 要: 通过DNA测序不仅可以更好地认识生命的本质, 了解生物的差异性、进化及发展史, 而且为重组DNA的研究提供了方向, 在疾病诊断及基因分型方面具有非常重要的实用价值。首先从发现DNA是遗传物质开始, 简要回顾了第一、二和三代测序技术的整个发展历程, 以及各自的优缺点和主要测序技术或平台;其次, 重点介绍了由PacBio公司开发的第三代测序典型技术——单分子实时测序技术 (SMRT) 和ONT纳米孔测序技术的原理、方法和技术流程, 并总结了第三代测序技术的应用领域, 包括基因组重测序、do novo测序、转录组研究、甲基化检测、与疾病相关的结构变异检测, 以及流行病学中病毒准种分析等应用;再次, 比较了三代测序技术在转录组测序和表观遗传学研究中的技术优势, 第三代测序技术在基因组重复区域或结构变异等研究领域具有非常明显的优势, 对多种疾病的研究意义重大, 已成为未来重要的精准诊断工具, 此外, 凭借对重要经济物种遗传信息的解析, 育种工作者通过建立基因与性状的关联, 对持有优良性状基因的个体进行人工选育, 大大缩短了育种年限;最后, 对第三代测序技术在医疗、农业、环境等领域的应用前景进行了展望。
关键词: 第三代测序; 单分子测序; 长读长; 结构变异;
Abstract: DNA sequencing not only helps us better to understand the nature of life, biological differences, different biological evolutions and developmental history of organisms, but also provides direction for recombinant DNA research, and has very important practical value in disease diagnosis and genotyping.Firstly, from the discovery that DNA is a genetic material, we briefly review the entire development process of the first, second and third generation sequencing technologies, their respective advantages and disadvantages, and the main sequencing technologies or platforms in each.Secondly, we focus on the third generation sequencing typical technology developed by PacBio, including single molecule Real time (SMRT) , Oxford Nanopore Technologies (ONT) , introduce the principles, methods and technical processes, and summarize the application areas of the third generation sequencing technology, including the genomic resequencing, do novo sequencing, transcriptome studies, methylation testing, disease-related structural variation testing and the application of viral quasispecies analysis in epidemiology.Thirdly, the advantages of three generations of sequencing techniques in transcriptome sequencing and epigenetics are compared, the third generation sequencing technology has obvious advantages in research fields, such as genomic repeat regions or structural variations, and very meaningful for the study of various diseases, it is considered to be an important and accurate diagnostic tool in the future.In addition, breeders can artificially select individuals by establishing associations between genes and traits by analyzing the genetic information of important economic species, which greatly shortens the breeding period.Finally, we prospect the future application of the third generation sequencing technology in medicine, agriculture and environment.
Keyword: the third generation sequencing; single molecule sequencing; long reads; structural variations;
自1953年Watson和Crick提出DNA分子双螺旋结构以来, 对遗传信息的解码一直是生命科学工作者的追求, 很多研究者开始了对DNA测序技术的探索。1975年, Sanger发明链终止法, 正式开启了一代测序的时代。1990年, 人类基因组计划正式启动;2000年, 人类基因组草图绘制完成, 生命科学步入基因组学时代。DNA测序技术的发展为人类探索自身和其他生命的奥秘提供了可能, 同时, 基因组学时代的来临对DNA测序技术也提出了更高的要求, 推动了DNA测序技术的不断进步。目前科学研究已进入了高通量测序时代, 从单一、局部的基因或基因片段的研究转变成了对整个基因组的研究, 在基因组从头测序和转录组测序中应用较广。近几年来, 以单分子实时测序为代表的第三代测序技术开始进入人们的视野, 该测序技术跨越了一、二代较短读长而直接对DNA单个分子进行测序的新一代测序平台应用日益广泛。作者简要回顾了第一、二和三代测序技术的整个发展历程、各自的优缺点及主要测序技术或平台, 重点介绍了由PacBio公司开发的第三代测序典型技术原理、方法和技术流程, 并总结了第三代测序技术的应用领域, 对第三代测序技术在医疗、农业、环境等领域的应用前景进行了展望。
1、 测序技术的发展历程
1.1、 一代测序
1975年, Sanger等在聚合酶作用下利用引物对模板DNA链的合成发明“加减法”对DNA进行测序[1], 随后引入双脱氧核苷三磷酸 (ddNTP) , 正式形成双脱氧链终止法[2], 该方法又被称为Sanger法, 后期很多测序技术都是基于该技术衍生的。1977年, Sanger报道了噬菌体φX174的DNA序列[3], 这是第一个被测序的基因组, 人类首次实现了对生物遗传信息的解码。同年, Maxam等[4]发明了化学降解法, 化学降解法不同于双脱氧链终止法, 对原DNA降解片段而非聚合酶合成片段进行测序, 避免了合成时引入的错误。但是化学降解法过程操作繁琐, 对有毒化学品和放射性同位素接触较多, 逐渐被双脱氧链终止法替代[5]。化学降解法、双脱氧链终止法及其衍生的测序技术统称为一代测序技术, 其中以双脱氧链终止法应用最为广泛, 故概念上提及一代测序也常被认为是Sanger测序。
双脱氧链终止法操作简便, 获得了研究者的广泛认可, 基于双脱氧链终止原理衍生了很多DNA测序技术, 如利用荧光标记代替放射性标记、采用自动成像系统检测的荧光自动测序技术[6]。随着科技的进步, 一些实验室也对双脱氧链终止法的自动化进行了研究, 并进行了商业化, 代表性的自动化测序仪制造商如ABI和Pharmacia-Amersham公司[7]。其中, ABI公司的Sanger测序仪应用最为广泛, 该平台利用毛细管电泳技术和荧光标记技术实现自动化。人类基因组计划 (Human Genome Project) 的大部分测序工作就是通过ABI Prism 3700完成[8], 现阶段, 研究者的Sanger测序需求大部分在ABI 3730系列平台上实现[9]。
以Sanger法为代表的一代测序技术测序读长长 (可达1 000 bp) , 准确率高 (可达99.999%) , 对生物学研究具有重要意义, 至今仍是基因测序的金标准。不可忽视的是, 一代测序的通量低、成本高, 限制了其大规模高通量的应用。
1.2、 二代测序
20世纪90年代, 在大规模基因组学发展需求下, 人们一直试图开发通量更高、成本更低的测序技术, 区分于以Sanger法为代表的一代测序技术, 后来发展的测序技术称为二代测序技术 (second-generation sequencing, SGS) 。二代测序技术诞生之初被称为下一代测序技术 (next-generation sequencing, NGS) , 发展势头迅猛, 其中以Roche公司的454技术, Illumina公司的Solexa、Hiseq技术, ABI公司的SoLiD技术, Helicos公司的HeliScope技术等为典型代表[10]。二代测序通量高, 成本大幅降低, 测序周期也大大缩短。
Roche公司的454技术是第一个商业化的二代测序平台, 初期被很多研究者使用, 454技术采用焦磷酸测序法, 可获得较长的测序读长 (可达400 bp) , 但是不能准确测量同聚物长度, 会引入插入和缺失错误。ABI公司的SoLiD技术利用连接酶法, 而非其他测序常用的聚合酶, 通过8碱基单链荧光探针与模板配对, 2个碱基确定一个荧光信号, 进行双次测序, 准确度高, 测序读长2×50 bp, 但是后续拼接较复杂。目前, 世界上使用量最大的二代测序仪是Illumina公司的Solexa和Hiseq平台, 核心原理都采用边合成边测序的方法[11], 该技术每次只添加1个dNTP的特点能够很好地解决同聚物长度的准确测量问题, 主要测序错误来源于碱基的替换, 错误率在1.0%~1.5%。Helicos公司的HeliScope技术[12]在二代测序中属于单分子测序, 测序前不进行PCR扩增, 利用聚合酶将荧光标记单核苷酸加到引物上进行测序, 不进行DNA扩增避免了扩增时引入的错误和偏好性, 但是读长较短 (25~30 bp) , 导致拼接困难、质量低, 仪器成本高, 并未大规模应用。
另外, 利用半导体芯片技术的Ion Torrent测序也可被归到二代测序的范畴。Ion Torrent测序利用布满约120万个小孔的高密度半导体芯片为载体, 一个小孔为一个测序反应池, 在DNA聚合酶作用下, 核苷酸聚合到DNA延伸链上, 核苷酸聚合释放出氢离子, 引起反应池内pH改变, 反应池内的场效应晶体管传感器捕获离子信号, 离子信号转化为数字信号。该技术的发明人Jonathan Rothberg同时也是454测序技术的发明人, 其文库制备技术与454测序类似。与其他测序技术相比, Ion Torrent测序不需要昂贵的物理成像设备, 在芯片制造中利用广泛使用的集成电路制造技术和金属氧化物半导体, 实现了高密度高通量阵列的制作[13], 仪器设备成本和体积得以降低。另外, 测序速度快, 上机测序可在2~3.5 h完成, 目前芯片通量并不高, 适合小基因组和外显子测序。
与Sanger测序相比, 二代测序在通量和成本上具有无可比拟的优势, 在过去的十多年里, 二代测序技术迅猛发展, 凭借其低成本、高通量的优势在很多领域得到了应用, 在很多探索性研究中, 如对新物种基因组的de novo测序、目标区域或全基因组重测序、转录组测序、宏基因组测序、表观修饰测序等领域都取得了突破性的进展[14]。然而, 二代测序的缺点就在于较短读长不利于生物信息学分析、PCR扩增前后的DNA片段数目比例有差异, 会大大影响基因表达。这些缺点在一定程度上制约了第二代测序技术的应用和发展, 推进第三代单分子测序技术应运而生。
1.3、 三代测序
目前, 大部分二代测序技术需要先进行PCR扩增, 会引入碱基错配和序列偏好性, 使得一些DNA片段在扩增后发生了相对频率和丰度的改变, 从而影响了测序结果的准确性, 对DNA单分子的测序成为测序技术发展的方向。目前发展的三代测序 (third-generation sequencing, TGS) 的典型特点便是单分子检测。Helicos公司的HeliScope是第一个商业化的单分子测序平台, 但是其读长短, 存在很多技术限制, 并未得到广泛应用。目前获得较大认可的是PacBio公司开发的单分子实时测序 (single molecule Real-time, SMRT) 技术和ONT (Oxford Nanopore Technologies) 纳米孔单分子测序技术, 三代测序以长读长为优势, 实现了对碱基序列的实时读取, 测序时间得以缩短[15]。近十年, 三代测序技术已成为研究者的首选。自2014年开始, 应用三代测序技术发表文章的数量已赶超应用二代测序技术发表文章的数量, 且在2018年, 前者更是达到后者的3倍之多 (图1) 。
1.3.1、 单分子实时测序技术
PacBio公司开发的SMRT技术是目前认可度较高的三代测序技术, 该技术也应用了边合成边测序的策略, 通过对模板链的复制获得序列信息。测序原理:芯片载体称之为SMRTCell, 待测DNA片段化后, 双链两端连接发夹接头形成闭合的环状单链模板, 称为SMRTbell (图1、2) [15];SMRTbell加到SMRTCell上后, 扩散进入测序单元, 称之为ZMW (zero-mode waveguide) , ZMW提供用于光检测的最小可用体积;单分子DNA聚合酶被固定在ZMW内, 捕获SMRTbell进行复制, 4色荧光标记的dNTPs (dATP、dCTP、dGTP、dTTP) 与模板配对, 根据激发产生的光脉冲识别碱基 (图3A) ;每个ZMW记录的连续光脉冲信号可被认为是连续碱基序列, 称为CLR (continuous long read) ;SMRTbell为环状, 测完一条DNA链后可以循环测互补链, 如果聚合酶的寿命足够长, 则两条链都能够在一个CLR内进行多次的测序, 称为pass;通过识别切除发夹接头, CLR可被分为多个subread, 同一个ZMW内subread间共有的序列称为环状共有序列 (circular consensus sequence, CCS) ;如果模板DNA太长则一个CLR内不能多次测序, 不能形成CCS, 只能输出单条subread;因为SMRT测序的实时性, 可以通过脉冲信号峰检测碱基修饰情况, 如甲基化 (图3B) 。
SMRT技术能够实现超长读长的关键是DNA聚合酶, 读长与酶的活性有关, 而酶的活性受激光对其造成损伤的影响。SMRT技术准确性的关键是如何将反应信号与周围强大荧光背景区分, 该技术利用ZMW原理实现:在芯片上设计比检测激光波长 (数百纳米) 小的ZMW (外径100多纳米) , 激光从底部打上去不能穿透小孔进入上方溶液区域, 能量被限制在小范围, 正好足够覆盖需要检测的部分, 所捕获的信号仅来自这个小反应区域, 孔外过多游离核苷酸单体依然留在黑暗中, 从而实现背景降到最低 (图2A) [16]。较之二代测序, SMRT测序速度很快, 每秒约10个dNTP, 但是通量低, 1个SMRT芯片池上有150 000 ZMW, 但是由于聚合酶未能在ZMW内固定或超过一条DNA分子进入ZMW, 只有35~70 000 ZMW可进行有效测序。SMRT测序的另一个缺点是CLR的错误率高达11%~15%, 但是不同于二代测序偏向性的错误, SMRT测序错误是随机的, 可以通过足够的测序次数纠正, 15次测序的CLR准确率超过99%。但是由于CLR总长度受聚合酶寿命的限制, 测序次数与CCS长度是相反的关系, 即CCS越长, 产生测序次数越少, 准确率更低, 反之亦然[15]。近年来SMRT测序在小型基因组从头测序和完整组装中已有良好应用, 并且已经或将在表观遗传学、转录组学、大型基因组组装等领域发挥其优势, 促进基因组学的研究。
图1 应用二代和三代测序技术研究发表文章的数量统计
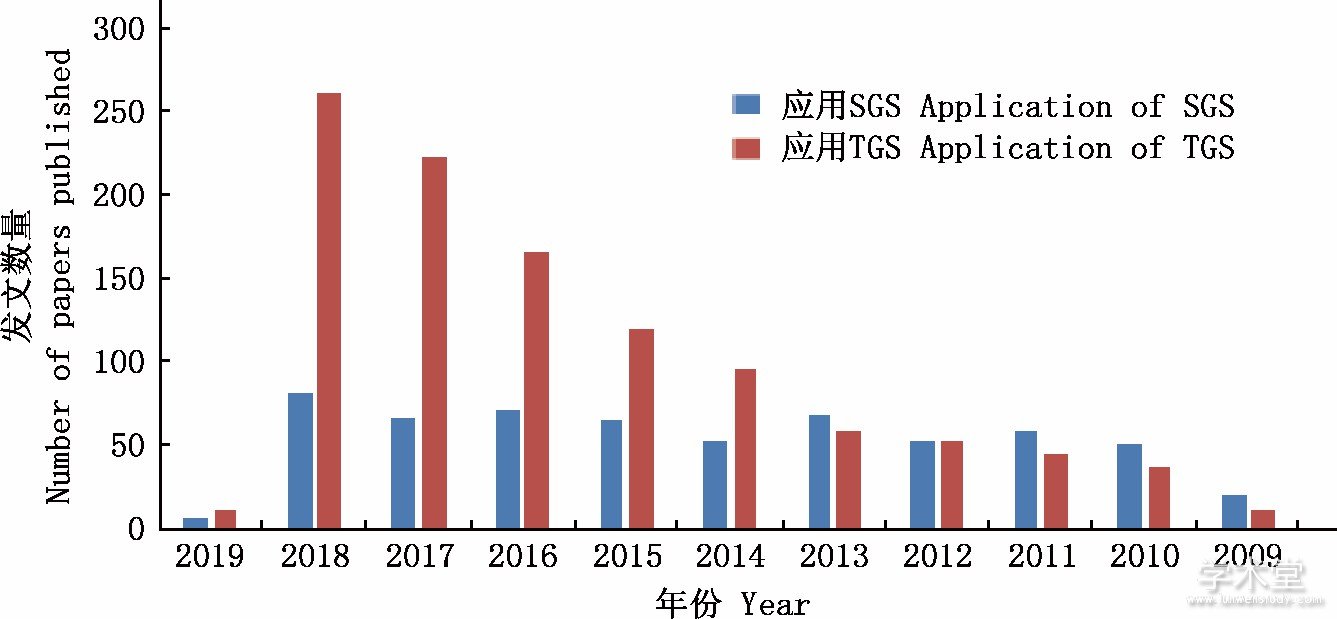
Fig.1 Statistics of the number of published papers applying second-generation and third-generation sequencing technology
数据来源于NCBI PubMed, 截止到2019年1月
The data are from NCBI PubMed as of January 2019
图2 SMRTbell示意图[15]
![图2 SMRTbell示意图[15]](http://www.xueshut.com/uploads/allimg/190925/36-1Z92515412cZ.jpg)
Fig.2 Schematic diagram of SMRTbell[15]
图3 SMRTCell碱基扩增 (A) 和碱基信号读取示意图 (B) [16]
![图3 SMRTCell碱基扩增 (A) 和碱基信号读取示意图 (B) [16]](http://www.xueshut.com/uploads/allimg/190925/36-1Z925154100U8.jpg)
Fig.3 Schematic diagram base amplification (A) and base signal reading of SMRTCell (B) [16]
1.3.2、 纳米孔单分子测序技术
三代测序技术的另一典型代表是ONT的纳米孔单分子测序技术, 是第二个商业化的三代测序平台。与其他技术不同, Nanopore基于电信号而非光信号。该技术的关键是设计一种特殊纳米孔, 在DNA分子通过纳米孔的过程中实现测序[17]。样本加到flow cell上, 当DNA分子通过纳米孔时, 电荷发生变化, 从而短暂影响通过纳米孔的电流强度, 每种碱基所影响的电流变化幅度不同, 通过检测电流变化鉴定碱基[18]。ONT测序的读长很长, 约几十甚至上百kb, 超过PacBio SMRT测序的读长;测序通量高, 起始DNA在测序过程中不被破坏;ONT测序样本制备简单便宜;数据可实时读取, 也可直接读取甲基化的胞嘧啶, 理论上也可检测RNA。ONT测序单碱基错误率高, 为随机错误, 可通过提高测序深度进而提高碱基准确率。2014年发布的ONT MinION (图4) , 实现了测序仪的便携化, 配备USB接口, 价格便宜, 测序速度快, 未来有望实现现场遗传信息的快速检测。纳米孔测序技术的样本处理简单, 无需DNA聚合酶或者连接酶, 也无需dNTPs, 测序成本十分低廉, 如果对该技术进一步完善和改进的话, 相信不久的将来也会变成核心技术[19]。
图4 MinION测序仪示意图[19]
![图4 MinION测序仪示意图[19]](http://www.xueshut.com/uploads/allimg/190925/36-1Z9251540395c.jpg)
Fig.4 Schematic diagram of MinION sequencer[19]
2、 三代测序技术的应用
2.1、 三代测序技术的优势
鉴于一代和二代测序存在依赖于模板扩增及序列读长限制等的缺点, 为了补充和进一步完善测序技术, 近几年研发出最新一代的测序方法——单分子测序技术 (single-molecule sequencing) , 主要包括Heli-cos公司的真正单分子测序技术 (true single-molecule sequencing, tSMSTM) 、Oxford Nanopore公司的单分子纳米孔测序技术 (single-molecule nanopore DNA sequencing) 、Pacific Biosciences (PacBio) 公司的SMRT等[20]。目前, 二代测序在基因组学领域已经取得了较好的工作基础, 二代和三代测序平台结合使用成为很多研究者的策略[15]。二代和三代测序各有所长, 二代测序读长短、通量高、准确度和性价比高, 而读长长、通量低、错误率高、单碱基成本高是三代测序的特点。利用二代测序高通量和准确度高的短读长片段对三代测序的长读长片段进行修正, 以降低三代测序的费用和错误率。三代测序的长读长则在序列比对、拼接和直接检测结构变异中具有显着的优势。此外, 利用三代测序进行基因组草图的gap填充, 不仅性价比高于Sanger测序, 而且能在一轮反应中填充超过2.5 kb的gap[21]。
2.2、 三代测序技术在基因组测序中的应用
凭借长读长优势, 三代测序在基因组de novo测序和重测序方向具有很大的应用空间, 尤其是对二代测序不能发现的重复基因组和结构变异区域的检测。利用三代测序实现了对包括模式生物在内的多个物种的基因组测序, 从简单的原核生物基因组到复杂的高等动植物基因组, 以及最受重视的人类基因组[22,23,24,25,26]。以三代测序在人基因组测序中的应用为例, Chaisson等[27]利用三代测序手段对人类单倍体基因组进行de novo组装, 填充了参考基因组GRCh37 164个gap中的50个, 并缩短了40个。在填充的50个gap中, 有39个包含高GC区的短串连重复序列, 而短串连重复序列通常不能被二代测序检测到, 另外此次测序发现了47 238个断点, 26 079个常染色体结构变异, 包括倒置、复杂插入和重复区域等。Shi等[26]对中国人个体HX1进行PacBio SMRT测序, 通过纳米通道微阵列构建物理图谱, de novo组装得到2.93 Gb的基因组和206 Mb的替代单倍型。拼装结果全部或部分填充274个 (28.4%) 人参考基因组GRCh38的N-gaps, 与GRCh38比较发现了12.8 Mb的HX1特有序列, 其中有4.1 Mb是没有被亚洲人基因组相关研究报道的。此外, 该研究对转录组的长读长测序也发现了未被GENCODE注释或未被短读长测序发现的稀有拼接基因。
通过长读长测序数据获得了对基因组成、结构变异和重复序列等描述更完整的图谱, 为群体遗传和进化研究提供了参考, 为个体实验室获得高质量复杂基因组提供了可能。Gordon等[23]对大猩猩 (Gorilla gorilla gorilla) 的基因组进行了测序拼接, 通过对1只大猩猩进行74.8×的PacBio SMRT全基因组测序, 拼接获得3.1 Gb的基因组, 为减少插入缺失错误和提高拼接准确性, 拼接时利用另外6只大猩猩的短读长测序基因组数据作为参考, 最终98.9%染色质基因组被拼接到1 854个contigs内, 大多数测序gap被填充, 87%缺失外显子和94%不完整基因组得到恢复。Guo等[28]利用包括Illumina、PacBio、Oxford Nanopore在内的多种测序技术对罂粟 (Papaver somniferum L.) 的基因组草图进行了构建, 并进一步通过共线性分析揭示了罂粟基因组中发生的基因重复、重排和融合事件, 这些基因组进化事件导致罂粟药用成分苄基异喹啉生物碱基因BIAs及其合成路径的形成。Tyson等[24]利用MinION测序分别对1株野生型和1株包含2个复杂重组的秀丽隐杆线虫 (Caenorhabditis elegans) 进行了基因组de novo测序, 在单个flow cell内获得了42×的深度, 对野生型秀丽隐杆线虫拼接的48个contigs覆盖了99%参考基因组, 并通过对重复区域的准确测序和对2个共提取细菌的完整基因组拼装使秀丽隐杆线虫的参考基因组扩大超过2 Mb, 对突变型的复杂重组阐释也更加深入。Ferrarni等[22]利用PacBio测序首次对蕨麻 (Potentilla micrantha) 的叶绿体基因组进行了测序, 1个contig就实现了对叶绿体基因组 (154 959 bp) 的覆盖, 且在高GC区无差异, 并对长的颠换重复区域实现了识别。Peng等[25]利用PacBio测序对小麦类作物致病菌 (Xanthomonas translucens) 的基因组和短读长测序难以拼接的转录激活因子样 (transcription-activator like, TAL) 效应物基因进行了测序拼接[25], 绘制了Xanthomonas translucens pv.undulosa strain XT4699基因组图谱和19个额外的Xanthomonas translucens菌株基因组草图, 为进一步的致病多样性和Xanthomonas translucens复合种群的宿主研究提供了参考, 菌株XT4699的TAL效应物在调控小麦宿主基因表达上起作用。Chin等[29]利用TGS测序及引入开源FALCON和FALCON-Unzip算法, 高度准确、连续地组装长读长序列数据, 由此提出了有关杂合样品的新的参考序列, 包括拟南芥F1杂种、一种广泛栽培的葡萄等。分阶段二倍体装配能够研究同源染色体之间的单倍型结构和杂合性, 包括鉴定编码序列内广泛的杂合结构变异, 该研究使得在非近交或重排杂合基因组组装的难题得以攻破。
2.3 、三代测序技术在结构变异检测中的应用
三代长读长测序在与很多疾病相关的结构变异 (structural variants, SVs) 研究中优势明显, 二代测序的短读长对结构变异区域覆盖有限, 而多达13%的人类基因组属于结构变异, 占变异碱基的大多数。Merker等[30]对常染色体显性遗传病Carney综合征进行研究, 利用Illumina HiSeq测序未能发现基因变异, 对病人进行低覆盖度的PacBio SMRT测序得到6 971个缺失和6 821个>50 bp的插入, 过滤掉无关变异并与基因编码外显子重叠, 鉴定出3个缺失和3个插入, 最终确定其中1个2 184 bp的杂合子缺失与Carney综合征相关基因PRKAR1A的第1个编码外显子重叠, RNA测序发现PRKAR1A基因表达量降低, 表明该缺失与基因致病有关, 该研究成功应用长读长测序第一次实现了对病人个体基因组致病变异的诊断。通过获得病人的短串连重复序列 (short tandem repeats, STRs) 扩展信息, 三代测序有望成为一种新的诊断方式, 这是短序列读长的二代测序不能实现的。Doi等[31]在个人基因组的长STRs研究中采用了组合测序方式, 对脊髓小脑性共济失调 (spinocerebellar ataxia 31, SCA31) 关联的STR进行测序, 发现了TGGAA和TAAAATAGAA的重复扩展决定了与SCR31相关的重复扩展不稳定性。Ishiura等[32]就家族性皮质肌阵挛性震颤伴癫痫 (familial cortical myoclonic tremor with epilepsy, FCMTE) 致病基因SAMD12的内含子4进行研究, 对变异等位基因结合细菌人工染色体组 (bacterial artificial chrome, BAC) 进行PacBio SMRT测序, 对病人的基因组进行MinION测序发现了SAMD12的五核苷酸序列TTTCA和TTTTA的异常重复扩展的致病变异, 而SAMD12无异常重复扩展的病人的另外2个致病基因TNRC6A和RAPGEF2的内含子则检出五核苷酸序列TTTCA和TTTTA的异常重复扩展。Zeng等[33]利用PacBio Sequel和Oxford Nanopore平台同样对中国人群的FCMTE致病基因SAMD12进行了鉴定, 确定了中国人群SAMD12的五核苷酸序列TTTCA和TTTTA的异常重复扩展的致病变异, 证实了Ishiura等[32]研究结果。值得注意的是, 该团队前期通过Array-CGH和全外显子测序、靶向捕获测序、全基因组测序等二代测序手段没有明确发现变异的复杂性。
值得一提的是, 长读长测序的准确度并不会因为序列存在多碱基重复而降低。人类脆性X智力缺陷1基因 (human fragile X mental retardation 1, FMR1) 正常有7~60个CGG重复, 发生倒置的为60~230个重复, 完全突变的为230个以上的重复。Loomis等[34]对完全突变的FMR1等位基因进行PacBio SMRT测序, 超过750个CGG重复并没有对PacBio测序结果造成干扰, 表明仅在聚合酶活性或1个CCS内subread数受到影响的情况下PacBio的测序结果才会受到影响, 而序列的重复结构不会影响测序结果。
2.4 、三代测序技术在转录组研究中的应用
三代测序的长读长在转录信息识别上更全面。PacBio的转录测序流程——Iso-Seq, 通过CCS方法将 mRNA形成环状分子循环多次测序, 可以不依赖参考基因组, 直接进行高达10 kb大小的转录组测序。应用该流程可对血细胞组分形成中的可变剪接进行研究, 该方法的实现对突变引起的可遗传疾病和血液肿瘤的作用机制阐述意义重大, 且在移植和再生医学的设计策略中具有应用价值。Haron等[35]应用该方法对20个人类器官和组织的多聚腺核苷酸RNA成分测序, 获得了476 000个CCS片段, 识别了14 000个拼接的GENCODE基因, 超过10%的比对结果为未注释的内含子结构。Wang等[36]利用PacBio SMRT测序对玉米转录组进行研究, 从6个组织中获得了111 151个转录本, 涵盖玉米RefGen_v3基因组注释中70%的基因, 其中, 57%转录本为新的、组织特异的或已知基因的亚型, 3%为新基因位点, 其他识别的转录本则优化了现有基因模型;6个组织的平均数据表明, 90%剪接位点来自于相应组织的短读长片段, 另外还发现了大量新的长链非编码RNA和融合转录本。Fleminh等[37]利用MinION进行转录组的研究, 对不同保存年限的大豆种子mRNA衰退进行比较发现, 相较于短时间保存 (2年) 的种子, 长时间保存 (23年) 的种子出现了转录片段化, mRNA出现非特异性的破坏, 长mRNA的片段化也更严重, 测序结果与自由基随机攻击碱基导致分子断裂的机制一致, 而短mRNA的高度完整并不代表种子没有死亡, 即短mRNA编码的蛋白并不足以保持活力。
通过三代长读长和二代短读长组合测序的策略, 可以对三代测序的转录组测序进行错误修正, 也可以提高基因分型的识别度和丰度估测。Tilgner等[38]对3个家庭的类淋巴母细胞转录组进行了PacBio和Illumina组合测序, 以产生和量化加强的个性化的基因组注释, 约711 000 CCS用来识别新亚型, 1亿的Illumina双端测序reads用来对个性化注释进行量化, 对于所有足量表达且<3 kb的基因, 这种方法产生的reads能够代表一个转录本的所有剪接位点。该方法提供了单核苷酸多态性 (SNPs) 的de novo策略, 能够提高RNA单倍体的推断准确率。Shi等[26]对中国人个体HX1进行Iso-Seq, 根据转录本大小分组建立文库 (1~2、2~3、3~5和>5 kb) , 对应测序数分别为11.2、8.4、6.9和5.2。利用短读长RNA-Seq数据对Iso-Seq数据进行错误修正, 在30 006个位点预测到58 383个高质量共识亚型。对于高表达的共识亚型, 在42个位点有57个亚型不与GENCODE中现有的转录本配对, 确认其中的一些拼接转录本具有超过2个的外显子。此类拼接转录本为灵长类特有, 在ENCODE所有9个细胞系的短读长RNA-Seq中未被检测到。另外, Iso-Seq确认11q13.4 (5个外显子) 和14q32.2 (4个外显子) 的2个稀有基因也未被RNA-Seq检测到。因此, 利用长度长测序方法结合生物学信息分析通过精确描述复杂的转录组能够辨别mRNA异构体, 且单分子测序的长度长测序技术发挥了重要作用。
2.5、 三代测序技术在甲基化检测中的应用
在细菌基因组中, 甲基化类型有N6-甲基腺嘌呤 (m6A) 、N4-甲基胞嘧啶 (m4C) 、5-甲基胞嘧啶 (m5C) [39]。在真核生物基因组中, 甲基化类型有N6-甲基腺嘌呤 (m6A) 、5-甲基胞嘧啶 (m5C) 、5-羟甲基胞嘧啶 (5hmC) 、5-甲酰胞嘧啶 (5fC) 和5-羧基胞嘧啶 (5caC) 。二代测序缺少简单的方法测定甲基化位置, 很多甲基化易被忽略[40]。目前胞嘧啶甲基化是研究最多的甲基化类型, 采用二代测序手段进行甲基化测序时, 需要先利用亚硫酸氢盐将未甲基化的胞嘧啶转化为尿嘧啶, 甲基化的胞嘧啶则保持不变, 通过检测碱基变化, 并与参考基因组比对, 确定甲基化位置[41]。该方法对参考基因组序列的准确度提出了较高的要求, 但由于亚硫酸氢盐处理过程不仅耗时, 而且反应过程会造成DNA降解, 因此该方法不能区分C、m5C和5hmC。
不同于二代测序需要预先利用亚硫酸氢盐还原, PacBio SMRT测序直接对自然状态下的甲基化碱基进行测序, 通过检测碱基在读长链上的通过时间, 即脉冲间隔持续时间 (inter-pulse durations, IPDs) , 确定甲基化状态。该技术通过对25种甲基化类型的特性区分, 可对DNA和RNA的甲基化修饰进行检测[39]。利用PacBio直接甲基化测序的原理, 可以同时实现de novo组装和表观遗传修饰识别, Satou等[42]通过对8株幽门螺旋杆菌 (Helicobacter pylori) 全基因组测序, de novo拼装得到了8个完整的contig, 而获得的甲基化信息则对与毒力因子相关的表观遗传修饰区域进行了识别。此外, ONT测序也可对甲基化碱基实现直接识别, 甲基修饰和无修饰碱基通过MinION纳米孔时, 引起的离子电流改变不同[43]。
利用三代测序手段, 不仅可对已有参考基因组或高覆盖度样本进行甲基化测序, 还可以对无参考基因组、新的甲基化类型和低覆盖度样本进行检测。Yao等[44]利用PacBio SMRT对人的DNA-6mA进行了研究, 确定了6mA甲基化在人类基因组中的存在, 发现了881 240个6mA修饰位点, 首次获得了中国人DNA-6mA修饰图谱。对甲基化图谱的分析发现, DNA-6mA在常染色体丰度较高, 在性染色体丰度较低, 且甲基化位点主要富集在外显子编码区。Murray等[39]利用PacBio SMRT对Geobacter metallireducens GS-15、Chromohalobacter salexigens、Vibrio breoganii 1C-10、Bacillus cereus ATCC 10987、Campylobacter jejuni subsp.jejuni 81-176和Campylobacter.jejuni NCTC 11168共6种细菌进行重测序, 发现了新的甲基化类型m6A、m4C。Beckmann等[41]在低覆盖度和高污染状态下检测到了m6A和m4C甲基化, 在3株混合菌株 (E.coli、G.metallireducens和C.salexigens) 的样本中发现了多个遗传修饰位点。因此, 通过SMRT测序系统不仅可以了解到基因表观遗传信息, 也为生物学和功能方面的应用开辟了新思路。
2.6、 三代测序技术在其他方面的应用
三代测序技术在其他领域的应用及研究也取得了非常重要的进展。三代测序技术可为人类流行病调查及菌种变型等研究提供资料, 从而采取相应的治疗手段。乙型肝炎病毒 (hepatitis B virus, HBV) 由于极高的复制率和在逆转录过程中的校对缺陷而具有高突变率, 产生的具有遗传异质性的变体被称为病毒准种 (QS) 。克隆测序 (clone-based sequencing, CBS) 作为检测QS复杂性和HBV多样性的“黄金标准”, 其检测成本高昂且过程复杂。三代测序技术与之相比, 具有更经济、操作简便且准确度高等特点, 在对未来HBV感染的临床管理中起到关键作用。三代测序技术在研究丙型肝炎病毒 (hepatitis C virus, HCV) 的抗性时提供了非常重要的资料。Takeda等[45]采用三代测序技术研究了HCV感染者服用抗病毒药 (DAA) 期间患者本身抗性相关变体 (RAV) 的动态, 发现在服用药物后非持续病毒学应答的患者中存在RAV的显性分离株, 且预先存在的RAV发展出多种耐药突变。治疗失败时的多种耐药性病毒克隆肯定源自HCV感染患者中预先存在的RAV的亚群, 从而得出在DAA治疗下, 筛选出的RAV成为具有多抗性的取代物。第三代测序平台带来的技术进步将明显扩大临床医生可用的诊断工具范围。但尽管如此, 这些诊断工具在临床实践中的使用仍处在前期的尝试阶段。三代测序技术除了可以为人类疾病的防控提供指导外, 还可以为研究动物RNA病毒的准种如H5亚型高致病性禽流感、猪牛口蹄疫、猪繁殖与呼吸综合征、鸡新城疫等提供试验资料[46,47]。在针对经典的N16961霍乱弧菌菌株的试验和模拟数据的单独控制组件中, 研究者利用三代测序技术获得了1 kb>14个拼接的试验数据及1 kb>8个拼接的模拟数据, 纠正了短读数据组装重叠群中的错误, 这项工作为快速微生物鉴定和全基因组装配提供了蓝图[48]。目前, 三代测序技术为人类流行病学和动物疾病的研究、诊断提供了资料, 方便采取相应的治疗手段, 虽然这些诊断方法在临床实践中的使用仍处在前期的尝试阶段, 但随着三代测序技术日趋完善, 将会在在组学研究中得到广泛应用。
3 、展 望
目前, 一代和二代测序技术渐趋成熟, 三代测序技术发展迅速, 根据研究需求选择组合的测序策略成为很多研究者的选择。在一代和二代测序的技术和数据积累基础上, 三代测序的长读长和单分子测序的优势毋庸置疑, 弥补了一代和二代测序在读长上的局限性, 避免了扩增引入的碱基偏好错误。目前, 以PacBio SMRT和ONT纳米孔测序技术为代表的三代测序技术在基因组学研究中应用日益增多, 研究领域涵盖基础科学、疾病诊疗、农业及环境等领域[49,50,51]。值得注意的是, 三代测序的代表平台之一——ONT MinION, 凭借快速测速和方便携带, 在快速检测场景如传染病、临床微生物和宏基因组学等研究中具有广阔前景[50]。目前三代测序的数据分析开发并不如二代测序完善, 且与小型基因组相比, 大型基因组在Overlapping的组装上需要更高的计算成本, 仍需开发更高效的计算方法[15]。
通过测序技术对遗传信息的解码和基因组数据库的构建, 人类不仅得以窥探生命的密码, 更能从基因层面对人类疾病进行检测甚至干预, 对其他物种的研究也更加深入, 对生态环境的研究更加透彻。目前, 利用测序技术对疾病相关基因组区域进行研究, 是精准医疗的一大研究方向[51]。三代测序在基因组重复区域或结构变异等研究领域优势明显, 而结构变异特性的研究对于包括癌症在内的很多疾病研究意义重大, 如与基因组重复区域或结构变异相关的遗传性罕见病[52]。利用三代测序技术不仅可以建立基因变异与疾病的关联, 更被越来越多研究者看好成为未来重要的精准诊断手段。此外, 基于基因信息的祖源研究, 人类对物种起源进化和选择有了更全面的认识。凭借对其他物种尤其是经济物种遗传信息的解析, 育种工作者通过建立基因与性状的关联, 对持有优良性状基因的个体进行人工选育, 大大缩短了育种进程。在生态环境研究方面, 通过对生物群体的遗传解码, 对于人与环境和谐发展将有新的认识。
参考文献
[1] SANGER F, COULSON A R.A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase[J].Journal of Molecular Biology, 1975, 94 (3) :441-448.
[2] SANGER F, NICKLEN S, COULSON A R.DNA sequencing with chain-terminating inhibitors[J].Proceedings of the National Academy of Sciences, 1977, 74 (12) :5463-5467.
[3] SANGER F, AIR G M, BARRELL B G, et al.Nucleotide sequence of bacteriophage phi X174 DNA[J].Nature, 1977, 265 (5596) :687-695.
[4] MAXAM A M, GILBERT W.A new method for sequencing DNA[J].Proceedings of the National Academy of Sciences, 1977, 74 (2) :560-564.
[5] SCHUSTER S C.Next-generation sequencing transforms today’s biology[J].Nature Methods, 2008, 5 (1) :16-18.
[6] 马文丽.基因测序实验技术[M].北京:化学工业出版社, 2012.MA W L.The Experimental Technology of Gene Sequencing[M].Beijing:Chemical Industry Press, 2012. (in Chinese)
[7] ANSORGE W J.Next-generation DNA sequencing techniques[J].New Biotechnology, 2009, 25 (4) :195-203.
[8] LANDER E S, LINTON L M, BIRRRN B, et al.Initial sequencing and analysis of the human genome[J].Nature, 2001, 409 (6822) :860-921.
[9] VENTER J C, ADAMS M D, MYERS E W, et al.The sequence of the human genome[J].Science, 2001, 291 (5507) :1304-1351.
[10] SHENDURE J, JI H.Next-generation DNA sequencing[J].Nature Biotechnology, 2008, 26 (10) :1135-1145.
[11] MARDIS E R.Next-generation DNA sequencing methods[J].Annual Review of Genomics and Human Genetics, 2008, 9:387-402.
[12] METZKER M L.Sequencing technologies the next generation[J].Nature Reviews Genetics, 2010, 11 (1) :31-46.
[13] ROTHBERG J M, HINZ W, REARICK T M, et al.An integrated semiconductor device enabling nonoptical genome sequencing[J].Nature, 2011, 475 (7356) :348-352.
[14] BUERMANS H P, DEN DUNEN J T.Next generation sequencing technology:Advances and applications[J].Biochimica et Biophysica Acta-Biomembranes, 2014, 1842 (10) :1932-1941.
[15] RHOADS A, AU K F.PacBio sequencing and its applications[J].Genomics Proteomics Bioinformatics, 2015, 13 (5) :278-289.
[16] EID J, FEHR A, GRAY J, et al.Real-time DNA sequencing from single polymerase molecules[J].Science, 2009, 323 (5910) :133-138.
[17] FENG Y, ZHANG Y, YING C, et al.Nanopore-based fourth-generation DNA sequencing technology[J].Genomics Proteomics Bioinformatics, 2015, 13 (1) :4-16.
[18] LU H, GIORDANO F, NING Z.Oxford Nanopore MinION sequencing and genome assembly[J].Genomics Proteomics Bioinformatics, 2016, 14 (5) :265-279.
[19] 乌日拉嘎, 徐海燕, 冯淑贞, 等.测序技术的研究进展及三代测序的应用[J].中国乳品工业, 2016, 44 (4) :33-37.WURILAGA, XU H Y, FENG S Z, et al.Research progress of sequencing technologies and the application of third generation sequencing[J].China Dairy Industry, 2016, 44 (4) :33-37. (in Chinese)
[20] MCCARTHY A.Third generation DNA sequencing:Pacific biosciences’ single molecule Real time technology[J].Chemistry & Biology, 2010, 17 (7) :675-676.
[21] ZHANG X, DAVENPORT K W, GU W, et al.Improving genome assemblies by sequencing PCR products with PacBio[J].Biotechniques, 2012, 53 (1) :61-62.
[22] FERRARINI M, MORETTO M, WARD J A, et al.An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome[J].BMC Genomics, 2013, 14 (1) :670
[23] GORDON D, HUDDLESTON J, CHAISSPN M J, et al.Long-read sequence assembly of the gorilla genome[J].Science, 2016, 352 (6281) :aae0344.
[24] TYSON J R, ONEIL N J, JAIN M, et al.MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome[J].Genome Research, 2018, 28:266-274.
[25] PENG Z, HU Y, XII J, et al.Long read and single molecule DNA sequencing simplifies genome assembly and TAL effector gene analysis of Xanthomonas translucens[J].BMC Genomics, 2016, 17 (1) :21.
[26] SHI L, GUO Y, DONG C, et al.Long-read sequencing and de novo assembly of a Chinese genome[J].Nature Communications, 2016, 7:12065.
[27] CHAISSON M J, HUDDLESTON J, DENNIS M Y, et al.Resolving the complexity of the human genome using single-molecule sequencing[J].Nature, 2015, 517 (7536) :608-611.
[28] GUO L, WINZER T, YANG X, et al.The opium poppy genome and morphinan production[J].Science, 2018, 362 (6412) :343-347.
[29] CHIN C, PELUSO P, SEDLAZECK F J, et al.Phased diploid genome assembly with single molecule Real-time sequencing[J].Nature Methods, 2016, 13:1050-1054.
[30] MERKER J D, WENGER A M, SNEDDON T, et al.Long-read genome sequencing identifies causal structural variation in a Mendelian disease[J].Genetics in Medicine, 2018, 20 (1) :159-163.
[31] DOI K, MONJO T, HOANG P H, et al.Rapid detection of expanded short tandem repeats in personal genomics using hybrid sequencing[J].Bioinformatics, 2014, 30 (6) :815-822.
[32] ISHIURA H.Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy[J].Nature Genetics, 2018, 50 (4) :581-590.
[33] ZENG S, ZHANG M Y, WANG X J, et al.Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy[J].Journal of Medical Genetics, 2019, 56 (4) :265-270.
[34] LOOMIS E W, EID J S, PELUSO P, et al.Sequencing the unsequenceable:Expanded CGG-repeat alleles of the fragile X gene[J].Genome Research, 2013, 23 (1) :121-128.
[35] HARON D, TILGNER H, GRUBRRT F, et al.A single-molecule long-read survey of the human transcriptome[J].Nature Biotechnology, 2013, 31 (11) :1009-1014.
[36] WANG B, TSENG E, REGULSKI M, et al.Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing[J].Nature Communications, 2016, 7:11708.
[37] FLEMINH M B, PATTERSON E L, REEVES P A, et al.Exploring the fate of mRNA in aging seeds:Protection, destruction, or slow decay[J].Journal of Experimental Botany, 2018, 69 (18) :4309-4321.
[38] TILGNER H, GRUBERT F, SHARON D, et al.Defining a personal, allele-specific, and single-molecule long-read transcriptome[J].Proceedings of the National Academy of Sciences, 2014, 111 (27) :9869-9874.
[39] MURRAY I A, CLARK T A, MORGAN R D, et al.The methylomes of six bacteria[J].Nucleic Acids Research, 2012, 40 (22) :11450-11462.
[40] FLUSBERG B A, WEBSTER D R, LEE J H, et al.Direct detection of DNA methylation during single-molecule, Real-time sequencing[J].Nature Methods, 2010, 7 (6) :461-465.
[41] BECKMANN N D, KARRI S, FANG G, et al.Detecting epigenetic motifs in low coverage and metagenomics settings[J].BMC Bioinformatics, 2014, 15 (Suppl 9) :S16.
[42] SATOU K, SHIROMA A, TERUYA K, et al.Complete genome sequences of eight Helicobacter pylori strains with different virulence factor genotypes and methylation profiles, isolated from patients with diverse gastrointestinal diseases on Okinawa Island, Japan, determined using PacBio single-molecule Real-time technology[J].Genome Announcement, 2014, 2 (2) :e00286-14
[43] WESCOE Z L, SCHREIBER J, AKESON M.Nanopores discriminate among five C5-cytosine variants in DNA[J].Journal of the American Chemical Society, 2014, 136 (47) :16582-16587.
[44] YAO B, CHENG Y, WANG Z, et al.N (6) -methyladenine DNA modification in the human genome[J].Molecular Cell, 2018, 71 (2) :306-318.
[45] TAKEDA H, UEDA Y, INUZUKA T, et al.Evolution of multi-drug resistant HCV clones from pre-existing resistant-associated variants during direct-acting antiviral therapy determined by third-generation sequencing[J].Scientific Reports, 2017, 7:45605.
[46] 崔治中.病毒准种多样性及其在免疫选择压作用下的演变[J].生命科学, 2013, 25 (9) :843-852.CUI Z Z.Multiplicity of viral quasi species and its evolution under selective pressure of antibody immunity[J].Chinese Bulletin of Life Sciences, 2013, 25 (9) :843-852. (in Chinese)
[47] BASHIR A, KLAMMER A A, ROBINS W P, et al.A hybrid approach for the automated finishing of bacterial genomes[J].Nature Biotechnology, 2012, 30 (7) :701-707.
[48] JAIN M, KOREN S, MIGA K H, et al.Nanopore sequencing and assembly of a human genome with ultra-long reads[J].Nature Biotechnology, 2018, 36 (4) :338-345.
[49] KOREN S, PHILLIPPY A M.One chromosome, one contig:Complete microbial genomes from long-read sequencing and assembly[J].Current Opinion in Microbiology, 2015, 23:110-120.
[50] EISENSTEIN M.An ace in the hole for DNA sequencing[J].Nature, 2017, 550 (7675) :285-288.
[51] SANCHIS-JUAN A, STEPHENS J, FRENCH C E, et al.Complex structural variants resolved by short-read and long-read whole genome sequencing in mendelian disorders[J].Genome Medicine, 2018, 10 (1) :95.
[52] COUNCIL N R.Toward Precision Medicine:Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease[M].Washington D.C.:National Academies Press, 2011.





