摘 要: 利用高效液相色谱法测定电解液中微量尿素的含量,分别研究了测试过程中流动相配比、流速以及干扰物质(OH-,NO2-,HCOO-,CH3COO-,NH3,CH3OH,CH3CH2OH等)对尿素检测的影响。通过改变流动相不同配比、流速,进而调节尿素的出峰时间、面积,可以发现当流动相配比为40∶60(乙腈∶水,V/V),流速为0.15 mL/min时,尿素出峰面积最大,且干扰物质与尿素的HPLC峰分离明显对尿素的检测无明显干扰。在最佳HPLC检测条件下,HPLC能够完成0~40 ppm范围内的低浓度尿素含量检测,线性相关系数为0.99。
关键词: 高效液相色谱法; 电合成; 尿素;
Abstract: The content of urea in the electrolyte was determined by high performance liquid chromatography, and the effects of different ratio of the mobile phase, flow rate, and interfering substances (OH-, NO2-, HCOO-, CH3COO-, NH3, CH3OH, CH3CH2OH, etc.) on the detection of urea were studied. The retention time and the peak area of urea could be changed by adjusting the composition and the flow rate of the mobile phase. We found that the peak area of urea could reach the maximum value when the composition and the flow rate of mobile phase were 40∶60 (CH3CN∶H2O, V/V) and 0.15 mL/min, respectively. And the HPLC peak of urea could be separated with interfering substances. HPLC could achieve the low concentration urea detection from 0 to 40 ppm under the optimized detection conditions of urea, the linear relationship was y = 171.4x, and the regression coefficient was 0.99.
Keyword: high performance liquid chromatography(HPLC); electrosynthesis; urea;
传统的尿素[1,2,3,4]合成方法是以液氨和二氧化为原料在高温高压的反应条件制备,该过程需要消耗约全球80%的氨以及全世界2%的能量[5,6],而采用可持续电化学的方式合成尿素有望克服这些缺点。目前,电合成的尿素主要利用显色法(如脲酶法和二乙酰一肟法)进行定性和定量分析[7,8,9,10,11]。但是,尿素电合成过程主要是通过在HCO3-和NO2-/NO3-混合电解液中持续鼓入CO2气体实现,这就导致在整个反应过程中势必会伴随着CO2电还原、水还原以及NO2-/NO3-还原等多种副反应,所产生的复杂物质如OH-,NO2-,HCOO-,CH3COO-,NH3,CH3OH,CH3CH2OH等难免会影响显色法分析准确度,容易获得假阳性的实验结果[12,13,14,15]。因此,亟待建立一种灵敏可靠、准确高效的微量尿素检测方法[16,17]。本文介绍一种采用高效液相色谱检测法(HPLC)测定电解液中尿素含量的方法。

1、实验部分
1.1、仪器与试剂
Cary 60紫外可见分光亮度计、1220 Infinity II高效液相色谱仪(安捷伦(中国)公司);
NaHCO3(纯度>99%),NaOH(纯度>97%),NaNO3(纯度>99%),NaNO2(纯度>99%),CH3CH2OH(纯度>99%),HCOONa(纯度>99%),CH3COONa(纯度>99%,),NH3?H2O(25%~28%水溶液),CH3OH(纯度>99%),尿素(纯度>99%);乙腈(色谱纯)。
1.2、 实验方法
1.2.1 、色谱条件
Luna@5mm NH2 Phenomenex(250 mm× 4.6 mm)色谱柱;流动相:乙腈和水;柱温25 ℃;进样量20 μL。
1.2.2、 尿素溶液紫外可见吸收光谱
高效液相色谱配备紫外可见吸收检测器。首先测定尿素溶液的紫外可见吸收图谱,然后根据最佳吸收波长确定HPLC的检测波长。
1.2.3、 流动相流速及配比优化
尿素电合成所用电解液一般为0.1 mol/L HCO3-和0.02 mol/L / 0.01 mol/L NO3- / NO2-混合电解液。为增加检测难度,验证HPLC在高盐条件下的检测能力,以0.2 mol/L NaHCO3与0.1 mol/L NaNO3混合液来模拟电解液。由于目前尿素电合成的产量一般在1 mg/L量级,以50 mg/L尿素为模拟产量。因此,整个检测条件的优化过程都以含50 mg/L尿素的0.2 mol/L NaHCO3与0.1 mol/L NaNO3混合溶液为模拟样品进行调试,综合考虑尿素的出峰面积大小、出峰时间和电解质的出峰位置干扰情况,确定出尿素的最佳HPLC检测条件。其中:1)流动相流速分别为:1.00,0.75,0.60,0.45,0.30,0.15 mL/min。2)流动相乙腈:水的比例分别为:10∶90,25∶75,40∶60,55∶45,70∶30 (V/V)。
1.2.4、共存物质的影响
尿素电合成过程中伴随着CO2还原、NO3-还原以及水还原反应,将产生多种物质,如CO2电还原的常见液相产物为HCOO-,CH3COO-,CH3OH,CH3CH2OH,NO3-电还原的常见液相产物为NH4+ 和NO2-,还有水还原产生的OH-,因此需确定这些干扰物质对尿素检测的影响。具体来说,用0.2 mol/L NaHCO3与0.1 mol/L NaNO3混合溶液分别配置上述物质溶液,控制它们的浓度均为50 mg/L。将上述各物质的HPLC出峰时间与尿素出峰时间相比较,如若与尿素出峰时间相同或者相近,即说明该物质在液相色谱中会干扰尿素的检测。
1.2.5、检测限及标准曲线
配置一系列浓度尿素标准液,用HPLC对其进行定量检测,确定对尿素浓度的检测限并绘制标准曲线。
2、结果与讨论
2.1、 检测波长
设置检测范围为190~400 nm。尿素在190 nm处具有强吸收。因此,HPLC紫外可见吸收光检测器选定190 nm作为尿素的检测波长。
2.2、 流动相配比和流速优化
图1 流动相乙腈与水不同配比下流速与尿素出峰时间图(a)和流速与尿素出峰面积图(b);尿素的HPLC图谱:不同流动相配比和1.00 mL/min流速(c);不同流动相配比和0.15 mL/min流速(d)
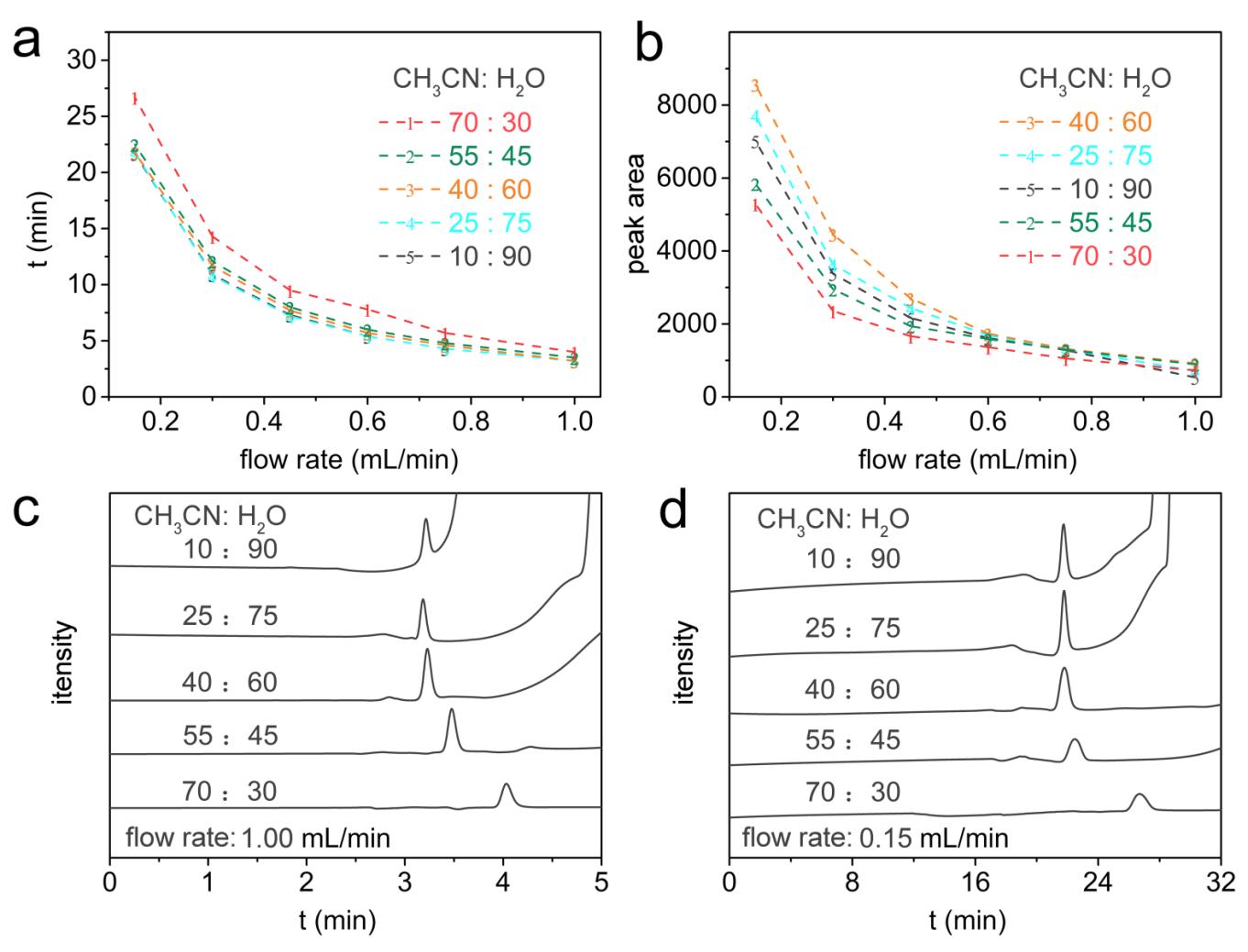
Fig. 1 The dependence of the retention time of urea(a) and the peak area of urea (b) on the flow rate of the mobile phase obtained under the different ratios of CH3CN to H2O; (c) HPLC spectra of urea obtained under the different ratios of mobile phase and the constant flow rate of 1.0 mL/min(c) and the constant flow rate of 0.15 mL/min (d)
图1a所示,流动相乙腈与水配比固定情况下,随着流速降低,尿素出峰时间延长,尿素出峰面积显着增加(图1b),尿素与电解质分离越明显(图1c和图1d);当流动相流速固定情况下,随着流动相中乙腈占比增加,尿素出峰时间有小幅度增加。可以发现,当流动相乙腈与水配比为40∶60(V/V),流速为0.15 mL/min时,尿素出峰面积最大(图1b),电解质与尿素的HPLC峰分离明显,此时,尿素出峰时间为21.9 min。
2.3 、其它物质干扰实验
采用2.2节,分别测试50 mg/L OH-,NO2-,HCOO-,CH3COO-,NH3,CH3OH,CH3CH2OH溶液。在尿素的出峰位置21.9 min处,均不存在这些物质HPLC峰,因此这些产物对尿素的检测无明显干扰。
2.4 、检测限和标准曲线
根据尿素浓度(X)及其HPLC峰面积(Y)对应值,由数据得出线性关系式为:Y = 171.4X,回归系数R2为0.99。因此HPLC能够完成0~40 mg/L范围内的低浓度尿素含量检测。检测限满足现阶段对尿素电合成微量产物浓度的检测要求。
3 、结论
建立了一种利用高效液相色谱法测定电解液中微量尿素含量的方法。室温下,采用氨基液相柱,尿素电合成副产物对该法尿素检测无干扰。尿素浓度与其HPLC峰面积在0~40 mg/L范围内呈现良好的线性关系。文中方法可实现电解液中尿素的定量检测,可满足现阶段对尿素电合成微量产物浓度的检测需要。
参考文献
[1] Erisman J W, Sutton M A, Galloway J, Klimont Z, Winiwarter W. Nat Geo Sci, 2008, 1(10): 636.
[2] Glibert P M, Harrison J, Heil C, Seitzinger S. Biogeochemistry, 2006, 77(3): 441.
[3] Rollinson A N, Jones J, Dupont V, Twigg M V. Eng Environ Sci, 2011, 4(4): 1216.
[4] Manaka Y, Nagatsuka Y, Motokura K. Sci Rep, 2020, 10(1): 2834.
[5] Giddey S, Badwal S P S, Kulkarni A. Int J Hydrogen Eng, 2013, 38(34): 14576.
[6] Barzagli F, Mani F, Peruzzini M.Green Chem, 2011, 13(5): 1267.
[7] Chen C, Zhu X, Wen X, Zhou Y, Zhou L, Li H, Tao L, Li Q, Du S, Liu T, Yan D, Xie C, Zou Y, Wang Y, Chen R, Huo J, Li Y, Cheng J, Su H, Zhao X, Cheng W, Liu Q, Lin H, Luo J, Chen J, Dong M, Cheng K, Li C, Wang S. Nat Chem, 2020, 12(8): 717
[8] Cao N, Quan Y, Guan A, Yang C, Ji Y, Zhang L, Zheng G. J Colloid Interface Sci, 2020, 577: 109.
[9] Shibata M, Yoshida K, Furuya N. Electroanal Chem, 1995, 387: 143
[10] Saravanakumar D, Song J, Lee S, Hur N H, Shin W. Chem Sus Chem, 2017, 10(20): 3999.
[11] Feng Y, Yang H, Zhang Y, Huang X, Li L, Cheng T, Shao Q. Nano Lett, 2020, 20(11): 8282.
[12] Figueiredo M C, Ledezma-Yanez I, Koper M T M. ACS Catal, 2016, 6(4): 2382.
[13] Meng N, Zhou W, Yu Y, Liu Y, Zhang B.ACS Catal, 2019, 9(12): 10983.
[14] Wang Y, Zhou W, Jia R, Yu Y, Zhang B. Angew Chem Int Ed Engl, 2020, 59(13): 5350.
[15] Jia R, Wang Y, Wang C, Ling Y, Yu Y, Zhang B. ACS Catal, 2020, 10(6): 3533.
[16] Rao L, Luo J, Bian C, Ning X, Li B. Chin J Anal Lab, 2020, 39(7): 796.
[17] Liu H, Xu B, Cao W, Dai X. Chin J Anal Lab, 2010, 29(6): 27.





